




Pedro Beltrao
@pedrobeltrao.bsky.social
Associate professor at ETH Zurich, studying the cellular consequences of genetic variation. Affiliated with the Swiss Institute of Bioinformatics and a part of the LOOP Zurich.
On my train back home from a two day visit to @embo.org The EMBL Heidelberg campus is always so beautiful in the autumn. Brings back memories of my PhD times.

October 29, 2025 at 6:50 PM
On my train back home from a two day visit to @embo.org The EMBL Heidelberg campus is always so beautiful in the autumn. Brings back memories of my PhD times.
With the Reading Assistant I tried to get it to act as reviewer and got a response that makes it look like they are trying their best to avoid this use case.

October 22, 2025 at 8:50 AM
With the Reading Assistant I tried to get it to act as reviewer and got a response that makes it look like they are trying their best to avoid this use case.
The Manuscript Adviser is more specific and currently provides feedback on tittle, abstract, missing references and overstated claims. The latter being the most interesting aspect so far. Similar to the gaps idea in www.qedscience.com but qed was much stronger on the one example I tested both on

October 22, 2025 at 8:50 AM
The Manuscript Adviser is more specific and currently provides feedback on tittle, abstract, missing references and overstated claims. The latter being the most interesting aspect so far. Similar to the gaps idea in www.qedscience.com but qed was much stronger on the one example I tested both on
To benchmark this Ricardo and @julianvangerwen.bsky.social have tested this on predicted structures if ubiquitinated proteins showing we get better accuracy when modelling with the covalently bound ubiquitin. We will continue to test this further.

October 16, 2025 at 10:56 AM
To benchmark this Ricardo and @julianvangerwen.bsky.social have tested this on predicted structures if ubiquitinated proteins showing we get better accuracy when modelling with the covalently bound ubiquitin. We will continue to test this further.
You may have noticed that AlphaFold3 does not allow covalent bonds between/within polymer chains (protein, DNA, RNA) but allows it for ligands. We tried to go around this limitation by simply treating one of residues/nucleic acids as a ligand.

October 16, 2025 at 10:47 AM
You may have noticed that AlphaFold3 does not allow covalent bonds between/within polymer chains (protein, DNA, RNA) but allows it for ligands. We tried to go around this limitation by simply treating one of residues/nucleic acids as a ligand.
Finally, we tested some examples by mutations in conserved sites and most excitingly, @klanglab.bsky.social 's lab used their new method of in vitro site-specific mono-ubiquitination to show that mono ubi ELAVL1 disrupts binding to RNA

October 9, 2025 at 7:07 AM
Finally, we tested some examples by mutations in conserved sites and most excitingly, @klanglab.bsky.social 's lab used their new method of in vitro site-specific mono-ubiquitination to show that mono ubi ELAVL1 disrupts binding to RNA
Julian built a ML model combining data on evolution, regulation, structures and MS evidence to predict "regulatory" sites. The model does well and suggests mechanisms for patient mutations. We tried to dig into some regulatory functions (activity, localization) in the manuscript

October 9, 2025 at 7:07 AM
Julian built a ML model combining data on evolution, regulation, structures and MS evidence to predict "regulatory" sites. The model does well and suggests mechanisms for patient mutations. We tried to dig into some regulatory functions (activity, localization) in the manuscript
We compiled a large dataset of 100 experiments where changes in ubiquitination were measured after perturbations. One really interesting finding from this was that highly conserved sites seem less linked with protein degradation and more with other signalling functions.

October 9, 2025 at 7:07 AM
We compiled a large dataset of 100 experiments where changes in ubiquitination were measured after perturbations. One really interesting finding from this was that highly conserved sites seem less linked with protein degradation and more with other signalling functions.
Eric Bennett's lab (UCSD) generated ubi data in different species that we combined with public data. While not many ubi-sites are highly conserved, these tend to be enriched in annotated functions. We find regulatory hotspot regions in domain families indicative of ancient regulation.

October 9, 2025 at 7:07 AM
Eric Bennett's lab (UCSD) generated ubi data in different species that we combined with public data. While not many ubi-sites are highly conserved, these tend to be enriched in annotated functions. We find regulatory hotspot regions in domain families indicative of ancient regulation.
We first needed a reliable catalog of ubiquitination sites (ubi-sites). Andy Jones's lab and Juan Antonio Vizcaino's @pride-ebi.bsky.social team reprocessed 11 datasets to derive 108,431 ubi-sites with controlled FDR. We then studied their conservation and regulation across conditions.

October 9, 2025 at 7:07 AM
We first needed a reliable catalog of ubiquitination sites (ubi-sites). Andy Jones's lab and Juan Antonio Vizcaino's @pride-ebi.bsky.social team reprocessed 11 datasets to derive 108,431 ubi-sites with controlled FDR. We then studied their conservation and regulation across conditions.
At the Basel Computational Biology meeting, Zuzanna Lottenbach from the Snijder lab in our institute @imsb-eth.bsky.social presenting their fantastic work on imaging studies of glioblastoma www.nature.com/articles/s41...

September 9, 2025 at 7:59 AM
At the Basel Computational Biology meeting, Zuzanna Lottenbach from the Snijder lab in our institute @imsb-eth.bsky.social presenting their fantastic work on imaging studies of glioblastoma www.nature.com/articles/s41...
The LLM built by the Swiss AI initiative (Apertus) has made the tech news but I have seen no benchmark comparisons. The internal benchmarks in their technical report that place Apertus around the level of Llama 3.1, great for a fist attempt and open model github.com/swiss-ai/ape...

September 4, 2025 at 7:30 AM
The LLM built by the Swiss AI initiative (Apertus) has made the tech news but I have seen no benchmark comparisons. The internal benchmarks in their technical report that place Apertus around the level of Llama 3.1, great for a fist attempt and open model github.com/swiss-ai/ape...
This looks like a great resource. One striking thing from this is that they go from 100 billion predicted protein sequences to 3 billion when clustered at 90% identify which tell us also how we keep sequencing the same species over and over again.

September 3, 2025 at 9:12 AM
This looks like a great resource. One striking thing from this is that they go from 100 billion predicted protein sequences to 3 billion when clustered at 90% identify which tell us also how we keep sequencing the same species over and over again.
Unexpectedly, @jurgjn.bsky.social found that running Alphafold3 predictions for protein interactions can yield ipTM scores that are more predictive of true interactions when run in pools of proteins instead of pairwise predictions. Presumably, this reflects some sort of "competition effect".
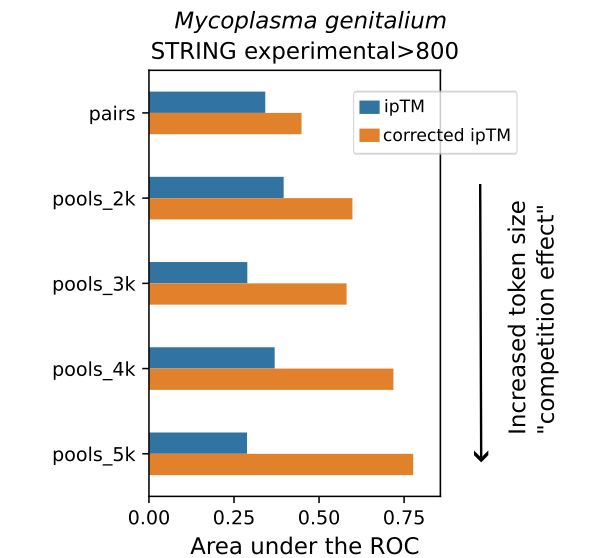
July 22, 2025 at 2:13 PM
Unexpectedly, @jurgjn.bsky.social found that running Alphafold3 predictions for protein interactions can yield ipTM scores that are more predictive of true interactions when run in pools of proteins instead of pairwise predictions. Presumably, this reflects some sort of "competition effect".
Lidia Gomez-Lucas in the @ivarssonlab.bsky.social tested some of the predicted human and viral peptide interfaces with 5 out of 6 human and 3 out of 6 viral interfaces validated. The lower success rate for the viral interfaces is likely to be real.
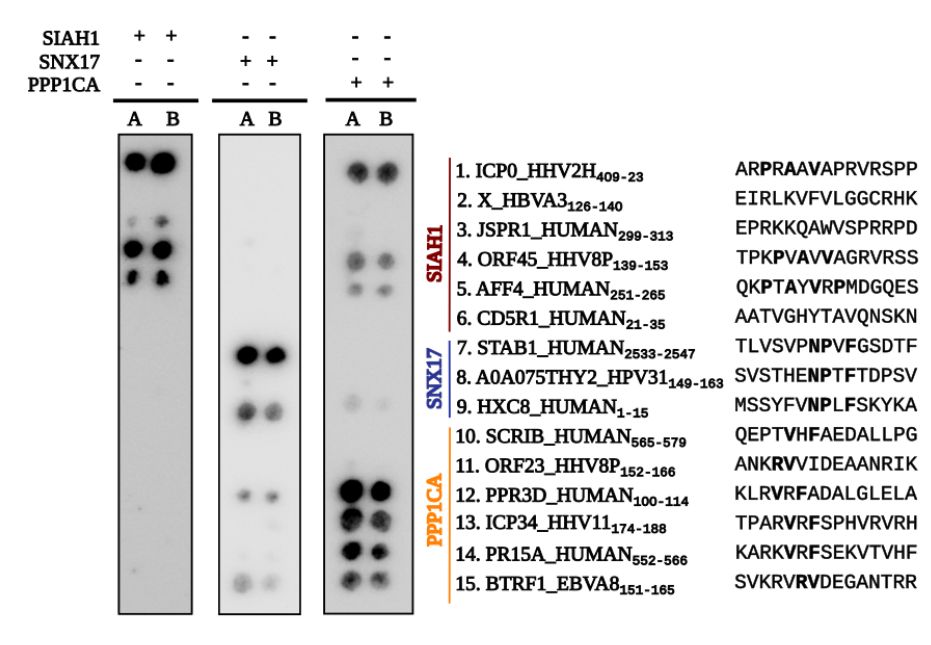
June 9, 2025 at 6:58 AM
Lidia Gomez-Lucas in the @ivarssonlab.bsky.social tested some of the predicted human and viral peptide interfaces with 5 out of 6 human and 3 out of 6 viral interfaces validated. The lower success rate for the viral interfaces is likely to be real.
we then compared predicted host-host and host-pathogen interfaces to quantify the degree and molecular mechanisms of mimicry. About half of the pathogen interfaces overlap with predicted host interfaces and linear-motif binding is the major mode of inferred mimicry.

June 9, 2025 at 6:58 AM
we then compared predicted host-host and host-pathogen interfaces to quantify the degree and molecular mechanisms of mimicry. About half of the pathogen interfaces overlap with predicted host interfaces and linear-motif binding is the major mode of inferred mimicry.
Out of 6,782 known host-pathogen interactions, 803 had higher model confidence. when the same human protein is targeted by more than 1 pathogen, we find they are typically (110 out of 138) predicted to bind the same interface, suggestive of convergent evolution. (see discussion for caveats)

June 9, 2025 at 6:58 AM
Out of 6,782 known host-pathogen interactions, 803 had higher model confidence. when the same human protein is targeted by more than 1 pathogen, we find they are typically (110 out of 138) predicted to bind the same interface, suggestive of convergent evolution. (see discussion for caveats)
we tested AF2 and AF3, seeing fairly similar (modest) performance - 30-35% "successful" models for structures deposited after the training cut-off period (no templates allowed). However, the ipTM score encouragingly enriches for successful models.

June 9, 2025 at 6:58 AM
we tested AF2 and AF3, seeing fairly similar (modest) performance - 30-35% "successful" models for structures deposited after the training cut-off period (no templates allowed). However, the ipTM score encouragingly enriches for successful models.
The major application we focused on was combining the brain derived protein associations with public pull-down data and co-fractionation data generated at ETH to re-prioritize candidate disease genes in GWAS linked loci.
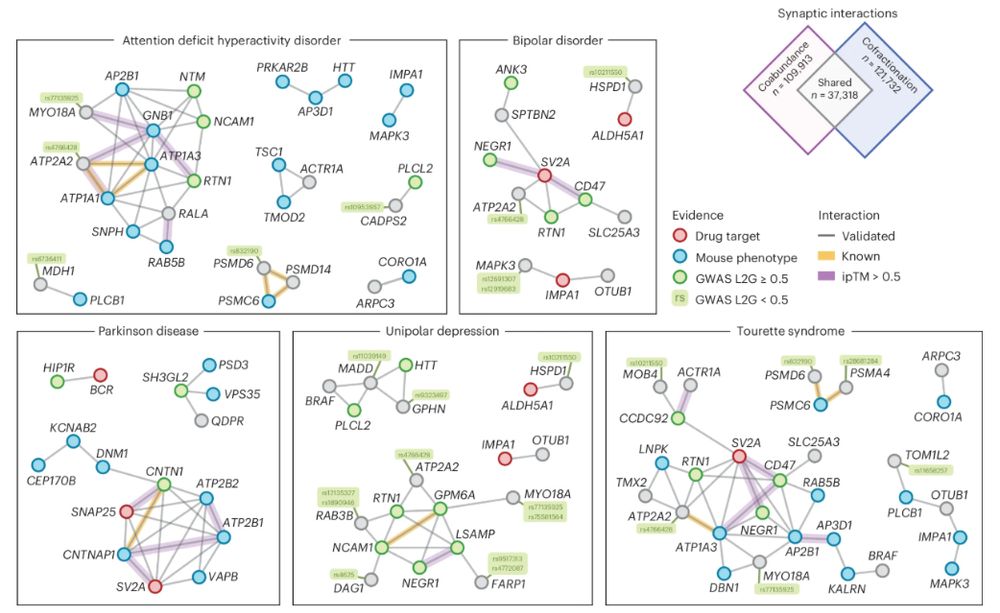
May 2, 2025 at 10:31 AM
The major application we focused on was combining the brain derived protein associations with public pull-down data and co-fractionation data generated at ETH to re-prioritize candidate disease genes in GWAS linked loci.
Given the limitations in accuracy, these associations are best used in system level analyses such as comparing gene sets and/or to combine with other data such as pull-downs. As an example, we can derive trait-to-trait and trait-to-compartment distances in a tissue specific manner.
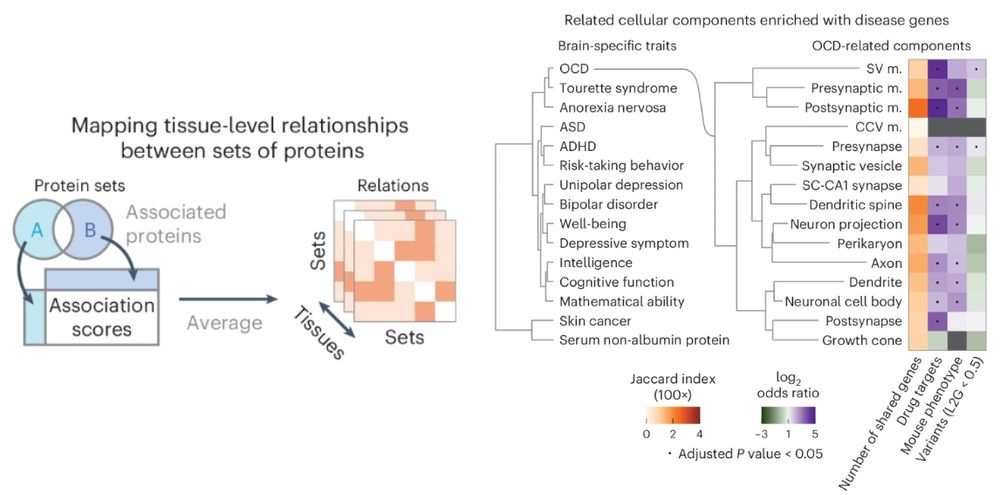
May 2, 2025 at 10:31 AM
Given the limitations in accuracy, these associations are best used in system level analyses such as comparing gene sets and/or to combine with other data such as pull-downs. As an example, we can derive trait-to-trait and trait-to-compartment distances in a tissue specific manner.
These predictions are not better than a general predictor like STRING that incorporates data of higher precision but we can show replication in a tissue specific manner. Surprisingly, we don't think that the majority of the difference in associations is explainable by tissue specific expression

May 2, 2025 at 10:31 AM
These predictions are not better than a general predictor like STRING that incorporates data of higher precision but we can show replication in a tissue specific manner. Surprisingly, we don't think that the majority of the difference in associations is explainable by tissue specific expression
Diederik compiled and harmonized, 7,811 public proteomic samples from 11 human tissues, confirming that co-expression networks are more accurately derived from protein abundance measurements (when compared to mRNA and even co-fractionation datasets).

May 2, 2025 at 10:31 AM
Diederik compiled and harmonized, 7,811 public proteomic samples from 11 human tissues, confirming that co-expression networks are more accurately derived from protein abundance measurements (when compared to mRNA and even co-fractionation datasets).
We are kicking off the Network Biology meeting at CSHL. I am looking forward to a few days of great science and seeing some familiar faces. We are starting with a keynote by Bonnie Berger people.csail.mit.edu/bab/

March 11, 2025 at 11:47 PM
We are kicking off the Network Biology meeting at CSHL. I am looking forward to a few days of great science and seeing some familiar faces. We are starting with a keynote by Bonnie Berger people.csail.mit.edu/bab/
The list of prioritized candidate genes was used to in a cohort of ciliopathy patients with unsolved genetic diagnoses leading to the identification of 3 patients with predicted pathogenic variants in CEP43 a novel candidate gene for human primary ciliopathies.

January 9, 2025 at 11:00 AM
The list of prioritized candidate genes was used to in a cohort of ciliopathy patients with unsolved genetic diagnoses leading to the identification of 3 patients with predicted pathogenic variants in CEP43 a novel candidate gene for human primary ciliopathies.
Expanding this idea to mouse gene deletion phenotypes can find the mouse phenotypes most closely linked to specific ciliopathies which in turn can be used to drive disease gene predictions, achieving good performance for many ciliopathies.


January 9, 2025 at 11:00 AM
Expanding this idea to mouse gene deletion phenotypes can find the mouse phenotypes most closely linked to specific ciliopathies which in turn can be used to drive disease gene predictions, achieving good performance for many ciliopathies.