



Antoine Limasset
@npmalfoy.bsky.social
CNRS researcher in bioinformatics
Lille, France (Bonsai team).
I develop efficient computational methods to analyze massive sequencing data, creating scalable tools for genomics, transcriptomics, and metagenomics.
https://malfoy.github.io/
Lille, France (Bonsai team).
I develop efficient computational methods to analyze massive sequencing data, creating scalable tools for genomics, transcriptomics, and metagenomics.
https://malfoy.github.io/
What the heck is that ???????

September 29, 2025 at 9:59 AM
What the heck is that ???????
This figure is extremely interesting, very surprising and exciting results from current state of sequencing technologies!

September 23, 2025 at 8:33 AM
This figure is extremely interesting, very surprising and exciting results from current state of sequencing technologies!
How is dev going ?
WELL....
WELL....

September 22, 2025 at 12:16 PM
How is dev going ?
WELL....
WELL....
This is a one to one Zoom call ....
WTF is Zoom client doing, mining Bitcoin ?
WTF is Zoom client doing, mining Bitcoin ?

July 15, 2025 at 2:32 PM
This is a one to one Zoom call ....
WTF is Zoom client doing, mining Bitcoin ?
WTF is Zoom client doing, mining Bitcoin ?
Indeed the size of the index is quite low and is quite robust with respect to the sequencing depth!

July 5, 2025 at 9:31 AM
Indeed the size of the index is quite low and is quite robust with respect to the sequencing depth!
We were even able to index Human ONT and HiFi datasets within hours using reasonable amount of memory and resulting in a surprisingly low index size!

July 5, 2025 at 9:31 AM
We were even able to index Human ONT and HiFi datasets within hours using reasonable amount of memory and resulting in a surprisingly low index size!
And it works pretty well!
We are able to be order of magnitude more resource-efficient than state of the art and globally scalable!
We are able to be order of magnitude more resource-efficient than state of the art and globally scalable!

July 5, 2025 at 9:31 AM
And it works pretty well!
We are able to be order of magnitude more resource-efficient than state of the art and globally scalable!
We are able to be order of magnitude more resource-efficient than state of the art and globally scalable!
We compared K2R with
movi (move index)
www.biorxiv.org/content/10.1...
SRC (MPHF and fingerprints) www.sciencedirect.com/science/arti...
Fulgor (MPHF and many CDBG tricks)
almob.biomedcentral.com/articles/10....
Themisto (spectral bwt) academic.oup.com/bioinformati...
movi (move index)
www.biorxiv.org/content/10.1...
SRC (MPHF and fingerprints) www.sciencedirect.com/science/arti...
Fulgor (MPHF and many CDBG tricks)
almob.biomedcentral.com/articles/10....
Themisto (spectral bwt) academic.oup.com/bioinformati...

July 5, 2025 at 9:31 AM
We compared K2R with
movi (move index)
www.biorxiv.org/content/10.1...
SRC (MPHF and fingerprints) www.sciencedirect.com/science/arti...
Fulgor (MPHF and many CDBG tricks)
almob.biomedcentral.com/articles/10....
Themisto (spectral bwt) academic.oup.com/bioinformati...
movi (move index)
www.biorxiv.org/content/10.1...
SRC (MPHF and fingerprints) www.sciencedirect.com/science/arti...
Fulgor (MPHF and many CDBG tricks)
almob.biomedcentral.com/articles/10....
Themisto (spectral bwt) academic.oup.com/bioinformati...
I won't spoil everything here as I want you to take a look at the paper but we borrow many techniques and tricks from colored de Bruijn graph to exploit theses properties and make the whole index efficient.
(Be not afraid by the figure)
(Be not afraid by the figure)

July 5, 2025 at 9:31 AM
I won't spoil everything here as I want you to take a look at the paper but we borrow many techniques and tricks from colored de Bruijn graph to exploit theses properties and make the whole index efficient.
(Be not afraid by the figure)
(Be not afraid by the figure)
1)We expect most kmers to occur in a very low amount of reads
2)Successive kmers are very likely to share the exact same origins
3)The amount of distinct "colors" ie read lists grow counterintuitively LINEARLY in an idealized scenario but also quite in practice
2)Successive kmers are very likely to share the exact same origins
3)The amount of distinct "colors" ie read lists grow counterintuitively LINEARLY in an idealized scenario but also quite in practice

July 5, 2025 at 9:31 AM
1)We expect most kmers to occur in a very low amount of reads
2)Successive kmers are very likely to share the exact same origins
3)The amount of distinct "colors" ie read lists grow counterintuitively LINEARLY in an idealized scenario but also quite in practice
2)Successive kmers are very likely to share the exact same origins
3)The amount of distinct "colors" ie read lists grow counterintuitively LINEARLY in an idealized scenario but also quite in practice
An interesting detail is that generic compressors don't handle reverse-complements.
To truly optimize compression, we need similar reads on the same strand!
Since this might affect strand-specific analyses, it's an optional feature.
To truly optimize compression, we need similar reads on the same strand!
Since this might affect strand-specific analyses, it's an optional feature.

July 3, 2025 at 10:53 AM
An interesting detail is that generic compressors don't handle reverse-complements.
To truly optimize compression, we need similar reads on the same strand!
Since this might affect strand-specific analyses, it's an optional feature.
To truly optimize compression, we need similar reads on the same strand!
Since this might affect strand-specific analyses, it's an optional feature.
We are extremely satisfied by these space results as we match state of the art performances in term of compressed size but this approach also boost decompression speed!

July 3, 2025 at 10:53 AM
We are extremely satisfied by these space results as we match state of the art performances in term of compressed size but this approach also boost decompression speed!
Despite the simplicity of the approach, it works extremely well!
Here a log scale plot of the compression of a HiFi dataset comparing xz,zstd,bz2,gz with various compression level with and without reordering.
State of the art tools performance are also included for reference.
Here a log scale plot of the compression of a HiFi dataset comparing xz,zstd,bz2,gz with various compression level with and without reordering.
State of the art tools performance are also included for reference.

July 3, 2025 at 10:53 AM
Despite the simplicity of the approach, it works extremely well!
Here a log scale plot of the compression of a HiFi dataset comparing xz,zstd,bz2,gz with various compression level with and without reordering.
State of the art tools performance are also included for reference.
Here a log scale plot of the compression of a HiFi dataset comparing xz,zstd,bz2,gz with various compression level with and without reordering.
State of the art tools performance are also included for reference.
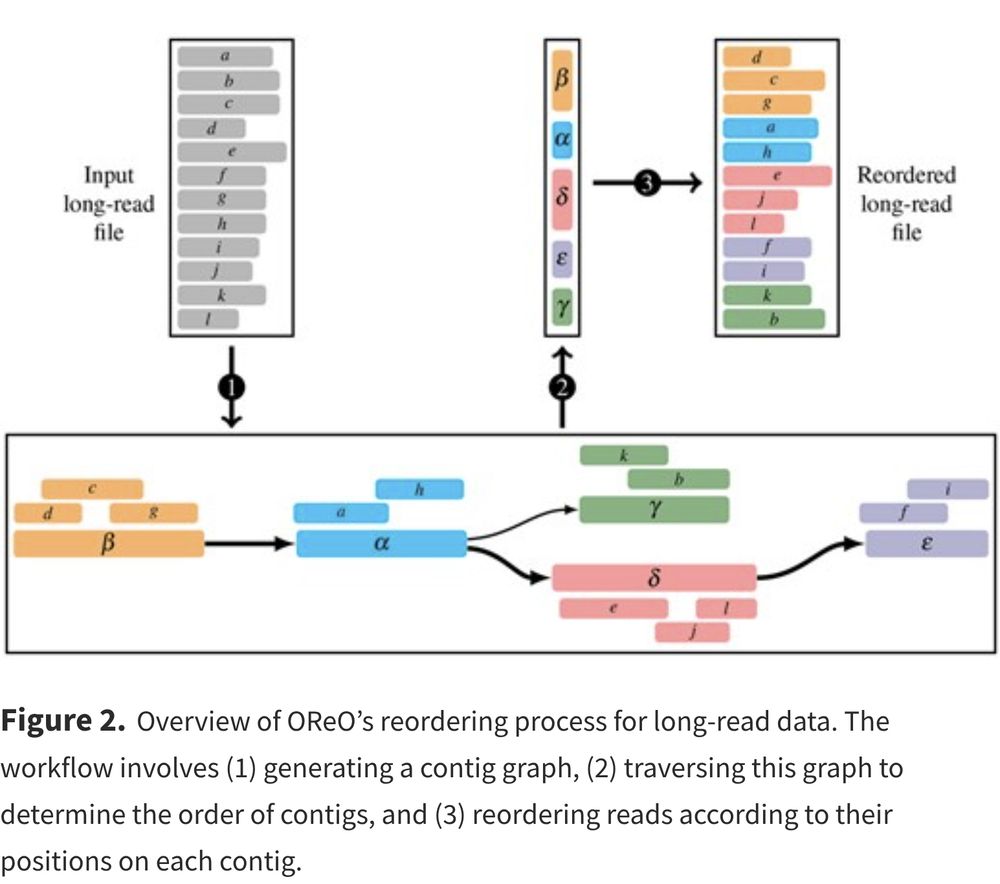
Since I want you to read the paper I won't spoil the whole method here :)
But the bird-eye view is that we are going to rely on a draft assembly graph to do so.
But the bird-eye view is that we are going to rely on a draft assembly graph to do so.
July 3, 2025 at 10:53 AM
Since I want you to read the paper I won't spoil the whole method here :)
But the bird-eye view is that we are going to rely on a draft assembly graph to do so.
But the bird-eye view is that we are going to rely on a draft assembly graph to do so.
In practice, we reorder the reads to "boost" the compression. After this step, we can use any generic compressor to efficiently compress this "equivalent" file. This file can then be decompressed with the corresponding decompressor, and it can also be efficiently compressed again.

July 3, 2025 at 10:53 AM
In practice, we reorder the reads to "boost" the compression. After this step, we can use any generic compressor to efficiently compress this "equivalent" file. This file can then be decompressed with the corresponding decompressor, and it can also be efficiently compressed again.
Overall K2Rmini is extremely efficient on various kind of searches, unitigs, contigs, long reads, short reads, whole genomes you name it!

July 2, 2025 at 1:00 PM
Overall K2Rmini is extremely efficient on various kind of searches, unitigs, contigs, long reads, short reads, whole genomes you name it!
A nice thing is that since we are based on mimizer, the larger the k the faster the search is!

July 2, 2025 at 1:00 PM
A nice thing is that since we are based on mimizer, the larger the k the faster the search is!
Of course we are still not optimal but even with a low amount of threads, we are able to get pretty close to the reading cost lower bound!
We mean need even faster parser, minimizer iterator or higher density minimizer...
We mean need even faster parser, minimizer iterator or higher density minimizer...

July 2, 2025 at 1:00 PM
Of course we are still not optimal but even with a low amount of threads, we are able to get pretty close to the reading cost lower bound!
We mean need even faster parser, minimizer iterator or higher density minimizer...
We mean need even faster parser, minimizer iterator or higher density minimizer...
By counting only the minimizers,a process that requires several-fold fewer hash table checks, we can reject most unrelated sequences and highly reduce the total number of lookups.
Hence, K2Rmini is basically a accelerated version of back_to_sequence, bringing order of magnitude improvement!
Hence, K2Rmini is basically a accelerated version of back_to_sequence, bringing order of magnitude improvement!

July 2, 2025 at 1:00 PM
By counting only the minimizers,a process that requires several-fold fewer hash table checks, we can reject most unrelated sequences and highly reduce the total number of lookups.
Hence, K2Rmini is basically a accelerated version of back_to_sequence, bringing order of magnitude improvement!
Hence, K2Rmini is basically a accelerated version of back_to_sequence, bringing order of magnitude improvement!
One of the main challenge for sequence bioinformatics in the last decades is to be able to exploit the, exponentially growing, goldmine that is the Sequence Read Archive
www.ncbi.nlm.nih.gov/sra/docs/sra...
www.ncbi.nlm.nih.gov/sra/docs/sra...

July 2, 2025 at 1:00 PM
One of the main challenge for sequence bioinformatics in the last decades is to be able to exploit the, exponentially growing, goldmine that is the Sequence Read Archive
www.ncbi.nlm.nih.gov/sra/docs/sra...
www.ncbi.nlm.nih.gov/sra/docs/sra...
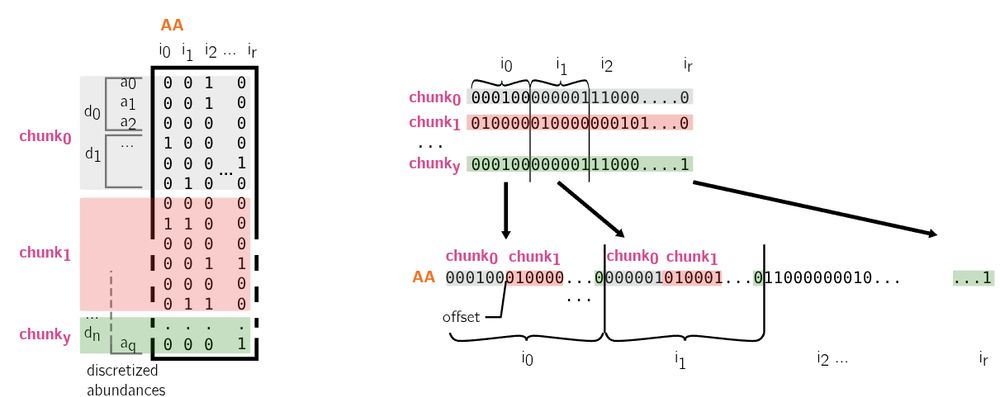
To keep low memory usage during construction, our index is inherently partitioned, meaning only a group of files are treated at a time. At the end of the process, all partitions are merged into a global index. This makes our index efficiently updatable as this is its vanilla construction method.
June 19, 2025 at 9:13 AM
To keep low memory usage during construction, our index is inherently partitioned, meaning only a group of files are treated at a time. At the end of the process, all partitions are merged into a global index. This makes our index efficiently updatable as this is its vanilla construction method.
The only downside of this approach is that the query time grows linearly with the amount of layers, meaning abundance levels times the number of datasets. While there is no free lunch, we still achieve very high throughput, processing 10k human transcripts (40 M bases) within a minute!

June 19, 2025 at 9:13 AM
The only downside of this approach is that the query time grows linearly with the amount of layers, meaning abundance levels times the number of datasets. While there is no free lunch, we still achieve very high throughput, processing 10k human transcripts (40 M bases) within a minute!
While having that many bloom filters could be expensive, these are pretty much empty.
Using sparse structures lets us have many large bloom filters at a very low cost in both time and memory.
Using sparse structures lets us have many large bloom filters at a very low cost in both time and memory.

June 19, 2025 at 9:13 AM
While having that many bloom filters could be expensive, these are pretty much empty.
Using sparse structures lets us have many large bloom filters at a very low cost in both time and memory.
Using sparse structures lets us have many large bloom filters at a very low cost in both time and memory.
Hence, we're introducing a log-based approximation designed to ensure a defined level of precision through the selection of a log base and a number of layers according to the abundance range to handle, minimum to maximum, and the desired precision.

June 19, 2025 at 9:13 AM
Hence, we're introducing a log-based approximation designed to ensure a defined level of precision through the selection of a log base and a number of layers according to the abundance range to handle, minimum to maximum, and the desired precision.
We insert kmers into Bloom filters. Following the needle approach, we use distinct Bloom filters to differentiate various abundance levels. If a kmer is found in a given Bloom filter, we can deduce both the dataset it appeared in and its corresponding abundance.

June 19, 2025 at 9:13 AM
We insert kmers into Bloom filters. Following the needle approach, we use distinct Bloom filters to differentiate various abundance levels. If a kmer is found in a given Bloom filter, we can deduce both the dataset it appeared in and its corresponding abundance.