




Nick Dimonaco
@nickdimonaco.bsky.social
PostDoc in 'Computational Biology' | Queen's University Belfast | Abersytwyth University | github.com/NickJD
Reposted by Nick Dimonaco

💾 any2fasta 0.8.1 is released!
The FASTA format is now 40 years old (Pearson & Lipman) and any2fasta makes it easy for your scripts and pipelines that accept FASTA to also accept other formats, even if compressed! eg. .gbk.gz
#bioinformatiocs #microbiology #genomcs
github.com/tseemann/any...
The FASTA format is now 40 years old (Pearson & Lipman) and any2fasta makes it easy for your scripts and pipelines that accept FASTA to also accept other formats, even if compressed! eg. .gbk.gz
#bioinformatiocs #microbiology #genomcs
github.com/tseemann/any...

Release Next time I'll try to be FASTA · tseemann/any2fasta
New features
Option -k is keep processing even when some inputs fail
option -g to include GBK version suffix
option s to strip desc from>id desc in ID lines
Support for PDB protein structure forma...
github.com
December 30, 2025 at 5:16 AM
💾 any2fasta 0.8.1 is released!
The FASTA format is now 40 years old (Pearson & Lipman) and any2fasta makes it easy for your scripts and pipelines that accept FASTA to also accept other formats, even if compressed! eg. .gbk.gz
#bioinformatiocs #microbiology #genomcs
github.com/tseemann/any...
The FASTA format is now 40 years old (Pearson & Lipman) and any2fasta makes it easy for your scripts and pipelines that accept FASTA to also accept other formats, even if compressed! eg. .gbk.gz
#bioinformatiocs #microbiology #genomcs
github.com/tseemann/any...
Reposted by Nick Dimonaco
💾 Prokka 1.15.6 is released!
This is the last major release of Prokka. But don't be sad, because @oschwengers.bsky.social already has an excellent replacement called Bakta you can migrate to.
#bioinformatics #microbiology #genomics
github.com/tseemann/pro...
This is the last major release of Prokka. But don't be sad, because @oschwengers.bsky.social already has an excellent replacement called Bakta you can migrate to.
#bioinformatics #microbiology #genomics
github.com/tseemann/pro...
Release Heading into the sunset · tseemann/prokka
The future
This is probably the last release of Prokka. I won't be making any code changes except bug fixes. I will update the databases occasionally. I strongly recommend you use Bakta by @oschwen...
github.com
December 15, 2025 at 9:09 PM
💾 Prokka 1.15.6 is released!
This is the last major release of Prokka. But don't be sad, because @oschwengers.bsky.social already has an excellent replacement called Bakta you can migrate to.
#bioinformatics #microbiology #genomics
github.com/tseemann/pro...
This is the last major release of Prokka. But don't be sad, because @oschwengers.bsky.social already has an excellent replacement called Bakta you can migrate to.
#bioinformatics #microbiology #genomics
github.com/tseemann/pro...
Reposted by Nick Dimonaco

Our new preprint, spelling out what a genomic-data-aware version control system could do to make #bioinformatics better for everyone.
It was really fun and hopeful, describing the potential for a better future with @nickdimonaco.bsky.social (QUB) and Martin Vickers (JIC). Although bioRxiv doesn't accept review paper preprints, here it is on Figshare figshare.com/articles/pre... 6/6
Genome Assemblies and Annotations Are Not Static and Need Support for Tracking Their Evolution
For the past 25 years, genomic data has been distributed in two key file formats, FASTA and GFF. These files are used across nearly all genomic analyses and encode both the data of genomic sequences ...
figshare.com
December 2, 2025 at 10:30 AM
Our new preprint, spelling out what a genomic-data-aware version control system could do to make #bioinformatics better for everyone.
Reposted by Nick Dimonaco
It was really fun and hopeful, describing the potential for a better future with @nickdimonaco.bsky.social (QUB) and Martin Vickers (JIC). Although bioRxiv doesn't accept review paper preprints, here it is on Figshare figshare.com/articles/pre... 6/6
Genome Assemblies and Annotations Are Not Static and Need Support for Tracking Their Evolution
For the past 25 years, genomic data has been distributed in two key file formats, FASTA and GFF. These files are used across nearly all genomic analyses and encode both the data of genomic sequences ...
figshare.com
December 1, 2025 at 7:07 PM
It was really fun and hopeful, describing the potential for a better future with @nickdimonaco.bsky.social (QUB) and Martin Vickers (JIC). Although bioRxiv doesn't accept review paper preprints, here it is on Figshare figshare.com/articles/pre... 6/6
Reposted by Nick Dimonaco
New blog post – A quick look at Roche's SBX
lh3.github.io/2025/09/11/a...
lh3.github.io/2025/09/11/a...

September 12, 2025 at 3:26 AM
New blog post – A quick look at Roche's SBX
lh3.github.io/2025/09/11/a...
lh3.github.io/2025/09/11/a...
Reposted by Nick Dimonaco

Sometimes you meet absolutely incredible bioinfo-magicians.
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...

Efficient sequence alignment against millions of prokaryotic genomes with LexicMap - Nature Biotechnology
LexicMap uses a fixed set of probes to efficiently query gene sequences for fast and low-memory alignment.
www.nature.com
September 10, 2025 at 9:12 AM
Sometimes you meet absolutely incredible bioinfo-magicians.
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
Reposted by Nick Dimonaco

The original circos plot? From the 1947-1948 Carnegie Yearbook, the page prior to McClintock's Mutable Loci in Maize paper.

August 16, 2025 at 4:24 PM
The original circos plot? From the 1947-1948 Carnegie Yearbook, the page prior to McClintock's Mutable Loci in Maize paper.
Reposted by Nick Dimonaco

“[The public] should understand that science ‘proves’ nothing, that conflicts of interest don’t automatically entail bias or fraud, that anomalous studies don’t falsify a whole body of literature and, above all, the public needs to understand that the scientist is human.”
doi.org/10.1007/s111...
doi.org/10.1007/s111...

August 15, 2025 at 6:50 AM
“[The public] should understand that science ‘proves’ nothing, that conflicts of interest don’t automatically entail bias or fraud, that anomalous studies don’t falsify a whole body of literature and, above all, the public needs to understand that the scientist is human.”
doi.org/10.1007/s111...
doi.org/10.1007/s111...
Reposted by Nick Dimonaco

If you ever find yourself needing evidence for ‘Plasmids are just as common in microbes without resistance genes,’ we’ve got you covered! Check our new paper, out today:
www.microbiologyresearch.org/content/jour...
www.microbiologyresearch.org/content/jour...

Plasmid prevalence is independent of antibiotic resistance in environmental Enterobacteriaceae
The rapid rise of antibiotic-resistant pathogens poses a critical threat to the treatment of infectious diseases. While the spread of antibiotic resistance genes (ARGs) via plasmid conjugation has bee...
www.microbiologyresearch.org
August 12, 2025 at 5:14 PM
If you ever find yourself needing evidence for ‘Plasmids are just as common in microbes without resistance genes,’ we’ve got you covered! Check our new paper, out today:
www.microbiologyresearch.org/content/jour...
www.microbiologyresearch.org/content/jour...
Reposted by Nick Dimonaco

🚨🚨 New paper in @narjournal.bsky.social! 🍾
Excludons are pairs of overlapping genes that block each other’s expression (basically, reverse operons).
We built a tool to identify them in bacterial genomes using transcriptomic data, in an awesome collab led by Iñigo Lasa and Álvaro San Martín.
👇
Excludons are pairs of overlapping genes that block each other’s expression (basically, reverse operons).
We built a tool to identify them in bacterial genomes using transcriptomic data, in an awesome collab led by Iñigo Lasa and Álvaro San Martín.
👇
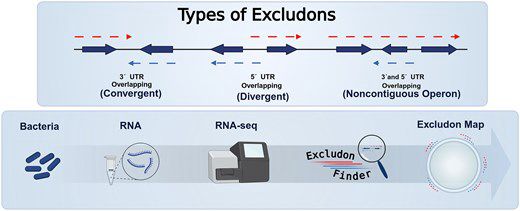
ExcludonFinder: mapping transcriptional overlaps between neighboring genes
Abstract. Bacteria regulate neighboring genes via overlapping transcription in untranslated regions (UTRs), forming excludons. This overlap leads to transc
academic.oup.com
July 25, 2025 at 9:26 PM
🚨🚨 New paper in @narjournal.bsky.social! 🍾
Excludons are pairs of overlapping genes that block each other’s expression (basically, reverse operons).
We built a tool to identify them in bacterial genomes using transcriptomic data, in an awesome collab led by Iñigo Lasa and Álvaro San Martín.
👇
Excludons are pairs of overlapping genes that block each other’s expression (basically, reverse operons).
We built a tool to identify them in bacterial genomes using transcriptomic data, in an awesome collab led by Iñigo Lasa and Álvaro San Martín.
👇
Reposted by Nick Dimonaco
"Our analysis identified 16 divergent and 165 convergent excludons in E. coli, as well as 10 divergent and 28 convergent excludons in S. aureus."
🚨🚨 New paper in @narjournal.bsky.social! 🍾
Excludons are pairs of overlapping genes that block each other’s expression (basically, reverse operons).
We built a tool to identify them in bacterial genomes using transcriptomic data, in an awesome collab led by Iñigo Lasa and Álvaro San Martín.
👇
Excludons are pairs of overlapping genes that block each other’s expression (basically, reverse operons).
We built a tool to identify them in bacterial genomes using transcriptomic data, in an awesome collab led by Iñigo Lasa and Álvaro San Martín.
👇
ExcludonFinder: mapping transcriptional overlaps between neighboring genes
Abstract. Bacteria regulate neighboring genes via overlapping transcription in untranslated regions (UTRs), forming excludons. This overlap leads to transc
academic.oup.com
July 25, 2025 at 9:31 PM
"Our analysis identified 16 divergent and 165 convergent excludons in E. coli, as well as 10 divergent and 28 convergent excludons in S. aureus."
www.biorxiv.org/content/10.1...
After spotting weird gene‐clustering and 'pangenome' quirks, I dug deep and found it’s not just one tool. It’s how we misuse and misassume how 'black-box' clustering and pangenome tools work.
#pangenomics #bioinformatics #geneclustering
After spotting weird gene‐clustering and 'pangenome' quirks, I dug deep and found it’s not just one tool. It’s how we misuse and misassume how 'black-box' clustering and pangenome tools work.
#pangenomics #bioinformatics #geneclustering

PyamilySeq: Exposing the fragility of conventional gene (re)clustering and pangenomic inference methods
Pangenomics, the identification of shared genes across a taxonomic range, is essential for understanding microbial genetic diversity. Yet, gene clustering and pangenome tools often operate as one-size...
www.biorxiv.org
July 22, 2025 at 10:42 AM
www.biorxiv.org/content/10.1...
After spotting weird gene‐clustering and 'pangenome' quirks, I dug deep and found it’s not just one tool. It’s how we misuse and misassume how 'black-box' clustering and pangenome tools work.
#pangenomics #bioinformatics #geneclustering
After spotting weird gene‐clustering and 'pangenome' quirks, I dug deep and found it’s not just one tool. It’s how we misuse and misassume how 'black-box' clustering and pangenome tools work.
#pangenomics #bioinformatics #geneclustering
Reposted by Nick Dimonaco

New paper with @rwheatley8.bsky.social and Cedric Lood
Actual title: Chromosomal capture of beneficial genes drives plasmids towards ecological redundancy.
Sensationalist title: Plasmids carry useless genes
academic.oup.com/ismej/articl...
Actual title: Chromosomal capture of beneficial genes drives plasmids towards ecological redundancy.
Sensationalist title: Plasmids carry useless genes
academic.oup.com/ismej/articl...
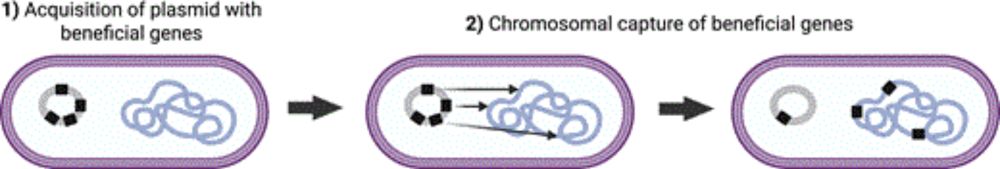
Chromosomal capture of beneficial genes drives plasmids toward ecological redundancy
Abstract. Plasmids are a ubiquitous feature of bacterial genomes, but the forces driving genes and phenotypes to become associated with plasmids are poorly
academic.oup.com
July 16, 2025 at 9:17 AM
New paper with @rwheatley8.bsky.social and Cedric Lood
Actual title: Chromosomal capture of beneficial genes drives plasmids towards ecological redundancy.
Sensationalist title: Plasmids carry useless genes
academic.oup.com/ismej/articl...
Actual title: Chromosomal capture of beneficial genes drives plasmids towards ecological redundancy.
Sensationalist title: Plasmids carry useless genes
academic.oup.com/ismej/articl...
Reposted by Nick Dimonaco

Absolutely amazing calibre of presentations in the first student competition at #CSM2025 today! I’m always so impressed by how much I learn from these, the next generation!
June 18, 2025 at 7:56 PM
Absolutely amazing calibre of presentations in the first student competition at #CSM2025 today! I’m always so impressed by how much I learn from these, the next generation!
Reposted by Nick Dimonaco
And ditto again in part 2 today! Everything from insect-microbe interactions to GM to remove AMR to dynamics in micro colonies. The future of Canadian microbiology is bright #CSM2025
Absolutely amazing calibre of presentations in the first student competition at #CSM2025 today! I’m always so impressed by how much I learn from these, the next generation!
June 19, 2025 at 9:25 PM
And ditto again in part 2 today! Everything from insect-microbe interactions to GM to remove AMR to dynamics in micro colonies. The future of Canadian microbiology is bright #CSM2025
Reposted by Nick Dimonaco

Oh, the irony of people using generative AI to "summarise" my papers 🫠
(here the real paper: link.springer.com/article/10.1...)
(here the real paper: link.springer.com/article/10.1...)

June 8, 2025 at 8:11 PM
Oh, the irony of people using generative AI to "summarise" my papers 🫠
(here the real paper: link.springer.com/article/10.1...)
(here the real paper: link.springer.com/article/10.1...)
Reposted by Nick Dimonaco

PyamilySeq: Exposing the fragility of conventional gene (re)clustering and pangenomic inference methods https://www.biorxiv.org/content/10.1101/2025.05.30.657108v1
June 3, 2025 at 5:51 AM
PyamilySeq: Exposing the fragility of conventional gene (re)clustering and pangenomic inference methods https://www.biorxiv.org/content/10.1101/2025.05.30.657108v1
Reposted by Nick Dimonaco

Using these proteins as a base, the magnificent Matt Schmitz developed a tool to study the prevalence of proteins within the human gut. This is InvestiGUT: github.com/Matt-Schmitz.... To allow its instant use we have made InvestiGUT accessible on our Galaxy instance: protologger.bi.denbi.de
GitHub - Matt-Schmitz/InvestiGUT: Lineage-specific microbial protein prediction facilitates exploration of protein ecology within the human gut.
Lineage-specific microbial protein prediction facilitates exploration of protein ecology within the human gut. - Matt-Schmitz/InvestiGUT
github.com
April 4, 2025 at 12:03 PM
Using these proteins as a base, the magnificent Matt Schmitz developed a tool to study the prevalence of proteins within the human gut. This is InvestiGUT: github.com/Matt-Schmitz.... To allow its instant use we have made InvestiGUT accessible on our Galaxy instance: protologger.bi.denbi.de
Reposted by Nick Dimonaco
Ever wondered if we are correctly predicting proteins from metagenomes? If so, we are happy to present our new work improving the prediction of proteins from the human gut microbiome: www.nature.com/articles/s41...! We corrected how each taxon's proteins are predicted and found WAY more proteins!

Lineage-specific microbial protein prediction enables large-scale exploration of protein ecology within the human gut - Nature Communications
Microbes within the gut vary in how they encode genes, both in terms of the genetic codes and gene structures, which are often unexplored in metagenomic analysis. Here, the authors develop a lineage-s...
www.nature.com
April 4, 2025 at 12:03 PM
Ever wondered if we are correctly predicting proteins from metagenomes? If so, we are happy to present our new work improving the prediction of proteins from the human gut microbiome: www.nature.com/articles/s41...! We corrected how each taxon's proteins are predicted and found WAY more proteins!
Reposted by Nick Dimonaco
2 days until this PDRA opportunity closes! Come join us as part of @mermanchester.bsky.social in (sometimes) sunny Manchester. #microsky #microbiome
🚨 The Whelan lab is hiring! 🚨 We are looking for a PDRA to join our lab at U of Manchester. We & collab Alvaro Mata (UofNottingham) are seeking someone interested in interdisciplinary techniques to est a hydrogel biofilm model to study microbe-pathogen interactions #Microsky #microbiome 🧪 🧬 💻

Research Associate: Academy of Medical Sciences (AMS) Springboard Award:Manchester
www.jobs.manchester.ac.uk
November 18, 2024 at 8:25 AM
2 days until this PDRA opportunity closes! Come join us as part of @mermanchester.bsky.social in (sometimes) sunny Manchester. #microsky #microbiome
Reposted by Nick Dimonaco
Here's our #GenomeInformatics24 poster on evaluating predictors of coding regions on raw reads (rather than on assembled genomes) doi.org/10.6084/m9.f... Why might you want to choose a predictor of coding regions for reads?
Predicting coding regions on unassembled reads, how hard can it be? - Genome Informatics 2024
This was presented at Genome Informatics 2024, Hinxton, UKBackground: Genome assembly and metagenome assembly is time consuming, space consuming, and often results in incomplete and/or inaccurate sequ...
doi.org
November 15, 2024 at 10:15 AM
Here's our #GenomeInformatics24 poster on evaluating predictors of coding regions on raw reads (rather than on assembled genomes) doi.org/10.6084/m9.f... Why might you want to choose a predictor of coding regions for reads?
Reposted by Nick Dimonaco
With rORForise (similar to ORForise) we aim to enumerate/tabulate all the possibilities, and to use these to let users know which predictors are more suitable for their task. It's still work in progress but here's early release code to keep an eye on github.com/NickJD/rORFo... (preprint coming soon)

GitHub - NickJD/rORForise: Read-based gene coverage evaluation
Read-based gene coverage evaluation. Contribute to NickJD/rORForise development by creating an account on GitHub.
github.com
November 15, 2024 at 10:15 AM
With rORForise (similar to ORForise) we aim to enumerate/tabulate all the possibilities, and to use these to let users know which predictors are more suitable for their task. It's still work in progress but here's early release code to keep an eye on github.com/NickJD/rORFo... (preprint coming soon)
Reposted by Nick Dimonaco
Here's a link to our #GenomeInformatics24 poster on "Finding the most diverse subset of proteins" that was presented yesterday. doi.org/10.6084/m9.f... Why do we want to find the most diverse set of proteins?
Finding the most diverse subset of proteins - Genome Informatics 2024
This was presented at Genome Informatics 2024, Hinxton, UK.Background: Knowledge of the natural diversity found among protein sequences is of use when engineering proteins, and also when understanding...
doi.org
November 14, 2024 at 10:07 AM
Here's a link to our #GenomeInformatics24 poster on "Finding the most diverse subset of proteins" that was presented yesterday. doi.org/10.6084/m9.f... Why do we want to find the most diverse set of proteins?