




Martin Fenk
@mfenk.bsky.social
PhD student @MPIIB Berlin | Exploring the captivating realm of microbial evolution within microbiomes #Microbiology #Evolution
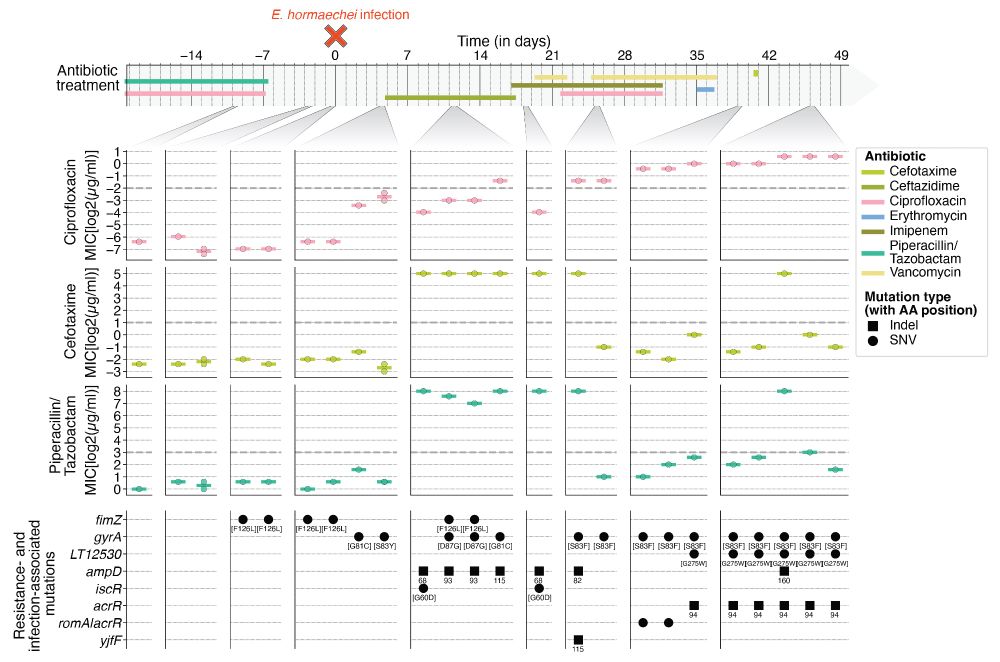
But why was the 𝘧𝘪𝘮𝘡 [F126L] mutant lost shortly after the HAI was cleared? Measuring the antibiotic resistance of isolates over time shows that the replacement was driven by multiple resistance-conferring mutations sweeping across the patient’s body, which were present until the end of the study.
October 27, 2025 at 9:25 AM
But why was the 𝘧𝘪𝘮𝘡 [F126L] mutant lost shortly after the HAI was cleared? Measuring the antibiotic resistance of isolates over time shows that the replacement was driven by multiple resistance-conferring mutations sweeping across the patient’s body, which were present until the end of the study.
Further, utilizing 𝘪𝘯 𝘷𝘪𝘷𝘰 survival assays with immunocompromised Relish 𝘋𝘳𝘰𝘴𝘰𝘱𝘩𝘪𝘭𝘢 𝘮𝘦𝘭𝘢𝘯𝘰𝘨𝘢𝘴𝘵𝘦𝘳, we observed an increase in its virulence leading to a more rapid decline in survival rates. This suggests that the 𝘧𝘪𝘮𝘡 [F126L] mutants’ fitness was elevated, potentially beneficial during pneumonia.

October 27, 2025 at 9:25 AM
Further, utilizing 𝘪𝘯 𝘷𝘪𝘷𝘰 survival assays with immunocompromised Relish 𝘋𝘳𝘰𝘴𝘰𝘱𝘩𝘪𝘭𝘢 𝘮𝘦𝘭𝘢𝘯𝘰𝘨𝘢𝘴𝘵𝘦𝘳, we observed an increase in its virulence leading to a more rapid decline in survival rates. This suggests that the 𝘧𝘪𝘮𝘡 [F126L] mutants’ fitness was elevated, potentially beneficial during pneumonia.
Does the 𝘧𝘪𝘮𝘡 [F126L] mutant convey a fitness effect? To answer this, we performed 𝘪𝘯 𝘷𝘪𝘵𝘳𝘰 assays and found this genotype translates into a hyperpilated phenotype with increased biofilm formation and adherence to lung epithelia.

October 27, 2025 at 9:25 AM
Does the 𝘧𝘪𝘮𝘡 [F126L] mutant convey a fitness effect? To answer this, we performed 𝘪𝘯 𝘷𝘪𝘵𝘳𝘰 assays and found this genotype translates into a hyperpilated phenotype with increased biofilm formation and adherence to lung epithelia.
During an 𝘌𝘯𝘵𝘦𝘳𝘰𝘣𝘢𝘤𝘵𝘦𝘳 𝘩𝘰𝘳𝘮𝘢𝘦𝘤𝘩𝘦𝘪 infection, we observe a short-lived, non-synonymous mutation in the fimbriae regulator gene 𝘧𝘪𝘮𝘡 [F126L] that is first observed in the gut, before being associated with HAI but subsequently swiftly replaced by repeated body-wide sweeps of independent genotypes.

October 27, 2025 at 9:25 AM
During an 𝘌𝘯𝘵𝘦𝘳𝘰𝘣𝘢𝘤𝘵𝘦𝘳 𝘩𝘰𝘳𝘮𝘢𝘦𝘤𝘩𝘦𝘪 infection, we observe a short-lived, non-synonymous mutation in the fimbriae regulator gene 𝘧𝘪𝘮𝘡 [F126L] that is first observed in the gut, before being associated with HAI but subsequently swiftly replaced by repeated body-wide sweeps of independent genotypes.
But were those pathogens acquired within the hospital or outside? Inferring the time to the most common recent ancestor reveals, that some lineages arose from a recent bottleneck during the hospital stay, while others have been colonizing the patient already before hospitalization.

October 27, 2025 at 9:25 AM
But were those pathogens acquired within the hospital or outside? Inferring the time to the most common recent ancestor reveals, that some lineages arose from a recent bottleneck during the hospital stay, while others have been colonizing the patient already before hospitalization.
While we have not seen one common reservoir microbiome niche for those pathogens, we identified that most pathogen strains are observable within the patient’s microbiome either before or within 6 h of HAI onset – pointing at the importance of the microbiome for HAI.

October 27, 2025 at 9:25 AM
While we have not seen one common reservoir microbiome niche for those pathogens, we identified that most pathogen strains are observable within the patient’s microbiome either before or within 6 h of HAI onset – pointing at the importance of the microbiome for HAI.
Of those, 3 patients developed HAIs by 9 different species. We performed culture-based sequencing of each pathogen of ≤10 isolates from the site of infection and ≤20 isolates/microbiome site/timepoint. This uncovered a closely related strain of the pathogens within the microbiome in 73% of cases.

October 27, 2025 at 9:25 AM
Of those, 3 patients developed HAIs by 9 different species. We performed culture-based sequencing of each pathogen of ≤10 isolates from the site of infection and ≤20 isolates/microbiome site/timepoint. This uncovered a closely related strain of the pathogens within the microbiome in 73% of cases.
To gain insights into the hidden evolutionary processes before, during and after HAI, we enrolled 13 critically ill patients and prospectively collected every week native nasal, oral, rectal and skin microbiome samples during their hospital stay.

October 27, 2025 at 9:25 AM
To gain insights into the hidden evolutionary processes before, during and after HAI, we enrolled 13 critically ill patients and prospectively collected every week native nasal, oral, rectal and skin microbiome samples during their hospital stay.