



VachaLab
@labvacha.bsky.social
Biological membranes, proteins. and their interactions at CEITEC MUNI (Brno, Czech Republic)
Second, we demonstrated how helical peptides can serve as sensors of membrane saturation: doi.org/10.1016/j.bp... Great work by Sushmita and Peter in collaboration with @javanainenm.bsky.social
Redirecting
doi.org
October 7, 2025 at 7:13 PM
Second, we demonstrated how helical peptides can serve as sensors of membrane saturation: doi.org/10.1016/j.bp... Great work by Sushmita and Peter in collaboration with @javanainenm.bsky.social
First, we showed the limitations of #Martini models in phospholipid flip-flop: doi.org/10.1021/acs.... Great work by Ondra, Ivo, and Lada!

Martini 3 Limitations in Phospholipid Flip-Flop
Phospholipid membranes serve as essential barriers in biological systems, and protein-mediated lipid flip-flop is a crucial process for lipid homeostasis in membranes, which is vital for various cellular functions. These phenomena can be studied using the Martini coarse-grained force field, a valuable tool for membrane simulations that balances computational efficiency with chemical accuracy while capturing key membrane properties. However, the accuracy of the newer Martini 3 force field in describing energetics of phospholipid flip-flop remains unknown. Here, we show dramatic differences in the free energy barriers of lipid flip-flop when simulated with Martini 3, Martini 2.2, and CHARMM36m force fields. Using umbrella sampling simulations of six phospholipids (POPC, DPPA, POPE, POPG, POPS, and DPTAP) in the POPC membrane, we demonstrate that Martini 3 predicts significantly lower flip-flop barriers compared to all-atom simulations and the older Martini 2.2 version, with particularly severe underestimation for the positively charged lipid (DPTAP). For DPTAP, we identified that altered Lennard-Jones parameters between choline and alkyl tail beads likely contribute to this discrepancy, as evidenced by our systematic parameter testing. These findings highlight limitations in the current Martini 3 parametrization that should be considered when studying processes involving molecular transport across membranes and suggest potential refinements to improve the model’s accuracy for such phenomena. To complete the picture, we also discuss the energetics of flip-flops of phospholipids with various lipid tail lengths (DLPC, DMPC, DPPC, DSPC) and lipid tail saturation (DOPC).
doi.org
October 7, 2025 at 7:13 PM
First, we showed the limitations of #Martini models in phospholipid flip-flop: doi.org/10.1021/acs.... Great work by Ondra, Ivo, and Lada!
Third, Tim and @denysbiriukov.bsky.social presented new collective variables to study the energetics of lipid membrane pores.
pubs.acs.org/doi/10.1021/...
Good job!
pubs.acs.org/doi/10.1021/...
Good job!
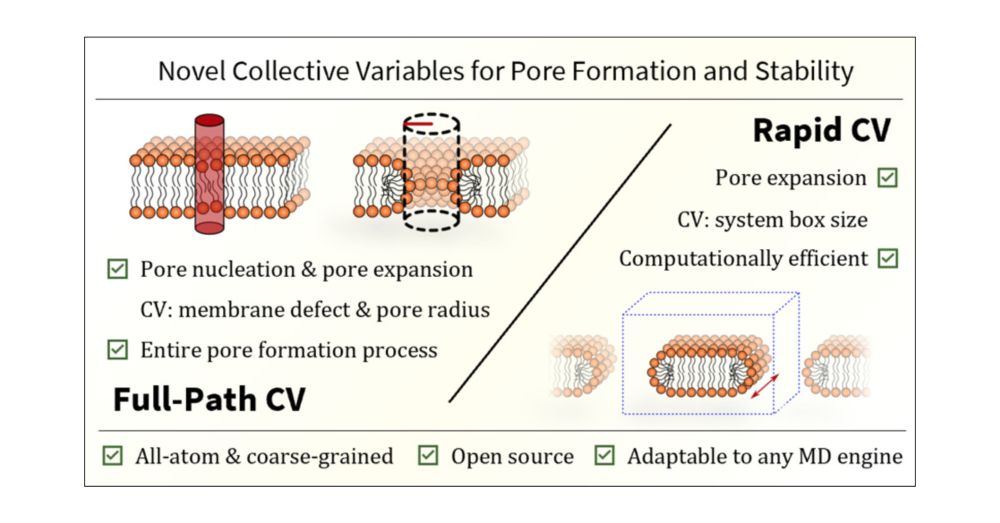
Free Energy of Membrane Pore Formation and Stability from Molecular Dynamics Simulations
Understanding the molecular mechanisms of pore formation is crucial for elucidating fundamental biological processes and developing therapeutic strategies, such as the design of drug delivery systems and antimicrobial agents. Although experimental methods can provide valuable information, they often lack the temporal and spatial resolution necessary to fully capture the dynamic stages of pore formation. In this study, we present two novel collective variables (CVs) designed to characterize membrane pore behavior, particularly its energetics, through molecular dynamics (MD) simulations. The first CV─termed Full-Path─effectively tracks both the nucleation and expansion phases of pore formation. The second CV─called Rapid─is tailored to accurately assess pore expansion in the limit of large pores, providing quick and reliable method for evaluating membrane line tension under various conditions. Our results clearly demonstrate that the line tension predictions from both our CVs are in excellent agreement. Moreover, these predictions align qualitatively with available experimental data. Specifically, they reflect higher line tension of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membranes containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS) lipids compared to pure POPC, the decrease in line tension of POPC vesicles as the 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) content increases, and higher line tension when ionic concentration is increased. Notably, these experimental trends are accurately captured only by the all-atom CHARMM36 and prosECCo75 force fields. In contrast, the all-atom Slipids force field, along with the coarse-grained Martini 2.2, Martini 2.2 polarizable, and Martini 3 models, show varying degrees of agreement with experiments. Our developed CVs can be adapted to various MD simulation engines for studying pore formation, with potential implications in membrane biophysics. They are also applicable to simulations involving external agents, offering an efficient alternative to existing methodologies.
pubs.acs.org
January 14, 2025 at 4:55 PM
Third, Tim and @denysbiriukov.bsky.social presented new collective variables to study the energetics of lipid membrane pores.
pubs.acs.org/doi/10.1021/...
Good job!
pubs.acs.org/doi/10.1021/...
Good job!
Second, Mehrnoosh showed how to "split" lipids to accelerate the sampling of all-atom lipid membranes.
pubs.acs.org/doi/10.1021/...
pubs.acs.org/doi/10.1021/...
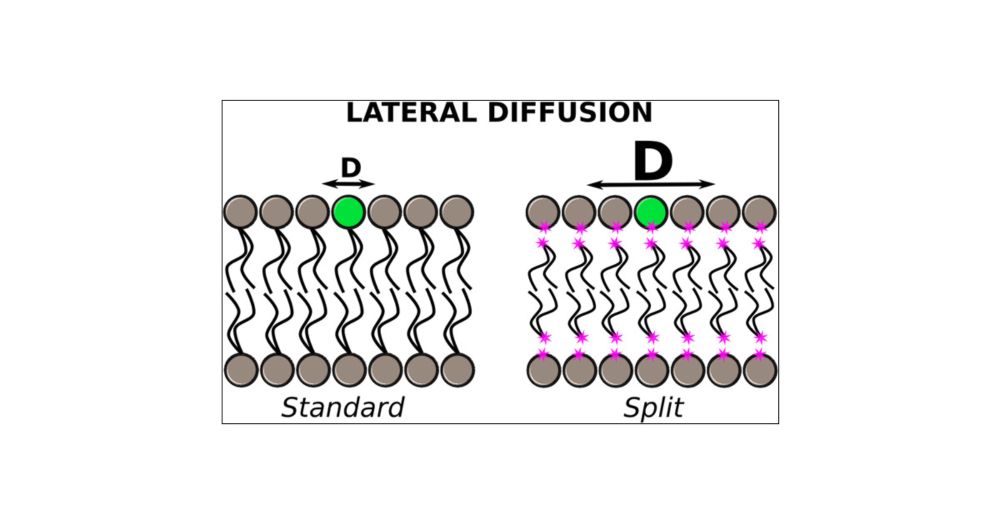
Split Membrane: A New Model to Accelerate All-Atom MD Simulation of Phospholipid Bilayers
All-atom molecular dynamics simulations are powerful tools for studying cell membranes and their interactions with proteins and other molecules. However, these processes occur on time scales determined by the diffusion rate of phospholipids, which are challenging to achieve in all-atom models. Here, we present a new all-atom model that accelerates lipid diffusion by splitting phospholipid molecules into head and tail groups. The bilayer structure is maintained by using external lateral potentials, which compensate for the lipid split. This split model enhances lateral lipid diffusion more than ten times, allowing faster and cheaper equilibration of large systems with different phospholipid types. The current model has been tested on membranes containing PSM, POPC, POPS, POPE, POPA, and cholesterol. We have also evaluated the interaction of the split model membranes with the Disheveled DEP domain and amphiphilic helix motif of the transcriptional repressor Opi1 as representative of peripheral proteins as well as the dimeric fragment of the epidermal growth factor receptor transmembrane domain and the Human A2A Adenosine of G protein-coupled receptors as representative of transmembrane proteins. The split model can predict the interaction sites of proteins and their preferred phospholipid type. Thus, the model could be used to identify lipid binding sites and equilibrate large membranes at an affordable computational cost.
pubs.acs.org
January 14, 2025 at 4:55 PM
Second, Mehrnoosh showed how to "split" lipids to accelerate the sampling of all-atom lipid membranes.
pubs.acs.org/doi/10.1021/...
pubs.acs.org/doi/10.1021/...