




John Lovell
@jotlovell.bsky.social
Helping to make genomics useful for crop improvement, ecology, evolutionary biology, and conservation
HudsonAlpha Genome Sequencing Center and DOE Joint Genome Institute
HudsonAlpha Genome Sequencing Center and DOE Joint Genome Institute
Very nice! Thanks for the link.
This is something you find when you dig deep enough. We've been looking for ways to harmonize annotations since we saw a similar pattern among pecan genomes in 2021 (buried in the SI tho). www.nature.com/articles/s41...
This is something you find when you dig deep enough. We've been looking for ways to harmonize annotations since we saw a similar pattern among pecan genomes in 2021 (buried in the SI tho). www.nature.com/articles/s41...

August 19, 2025 at 12:26 AM
Very nice! Thanks for the link.
This is something you find when you dig deep enough. We've been looking for ways to harmonize annotations since we saw a similar pattern among pecan genomes in 2021 (buried in the SI tho). www.nature.com/articles/s41...
This is something you find when you dig deep enough. We've been looking for ways to harmonize annotations since we saw a similar pattern among pecan genomes in 2021 (buried in the SI tho). www.nature.com/articles/s41...
Furthermore, even within fully present ('core') gene families we noticed a disturbing trend — identical sequence was not annotated with identical gene structures 20-50% of the time w/in annotation methods and 40-70% of the time btw methods
IGC-reannotation is not perfect, but reduces this to 5-15%
IGC-reannotation is not perfect, but reduces this to 5-15%

August 18, 2025 at 4:51 PM
Furthermore, even within fully present ('core') gene families we noticed a disturbing trend — identical sequence was not annotated with identical gene structures 20-50% of the time w/in annotation methods and 40-70% of the time btw methods
IGC-reannotation is not perfect, but reduces this to 5-15%
IGC-reannotation is not perfect, but reduces this to 5-15%
But what about within methods? Is using the same method enough to trust PAV? The answer here is less obvious, but method clearly matters.
Within two groups that annotated 7 and 23 soybean genomes there were 3x & 2x more PAVs than IGC — these pangenomes are not as 'open' as reported.
Within two groups that annotated 7 and 23 soybean genomes there were 3x & 2x more PAVs than IGC — these pangenomes are not as 'open' as reported.

August 18, 2025 at 4:51 PM
But what about within methods? Is using the same method enough to trust PAV? The answer here is less obvious, but method clearly matters.
Within two groups that annotated 7 and 23 soybean genomes there were 3x & 2x more PAVs than IGC — these pangenomes are not as 'open' as reported.
Within two groups that annotated 7 and 23 soybean genomes there were 3x & 2x more PAVs than IGC — these pangenomes are not as 'open' as reported.
In other words, while gene PAV similarity of IGC re-annotated genomes recapitulates known relatedness, clustering by original annotation PAV simply distinguished which consortium did the annotation (and did not evolutionary relationships): PAV across the original annotations is largely artifactual.

August 18, 2025 at 4:51 PM
In other words, while gene PAV similarity of IGC re-annotated genomes recapitulates known relatedness, clustering by original annotation PAV simply distinguished which consortium did the annotation (and did not evolutionary relationships): PAV across the original annotations is largely artifactual.
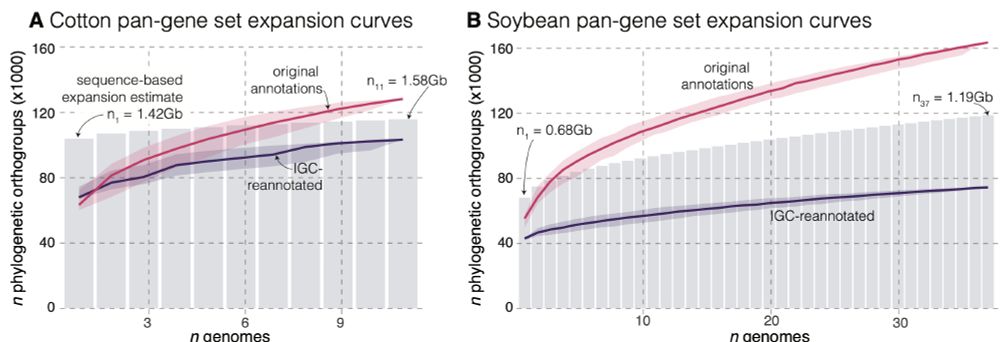
To develop a baseline, we re-annotated the genomes with exactly the same 'Integrated Gene Caller' (IGC) pipeline. IGC annotations had ⬆️ BUSCO and ⬇️ false positives, yet more than halved PAV%. Critically, assembly-based relatedness predicted PAV similarity from IGC but not original annotations.

August 18, 2025 at 4:51 PM
To develop a baseline, we re-annotated the genomes with exactly the same 'Integrated Gene Caller' (IGC) pipeline. IGC annotations had ⬆️ BUSCO and ⬇️ false positives, yet more than halved PAV%. Critically, assembly-based relatedness predicted PAV similarity from IGC but not original annotations.
We downloaded 'original' genome annotations directly from Soybase and Cottengen repos and calculated gene families from OrthoFinder. In both species there were WAY more PAVs than we expected: ~140k (86%) and ~90k (62%) of gene families were absent in ≥1 soybean and cotton genome respectively.

August 18, 2025 at 4:51 PM
We downloaded 'original' genome annotations directly from Soybase and Cottengen repos and calculated gene families from OrthoFinder. In both species there were WAY more PAVs than we expected: ~140k (86%) and ~90k (62%) of gene families were absent in ≥1 soybean and cotton genome respectively.
We first looked at how divergence time correlates with gene PAV in pairs of plant and animal genomes that were annotated with the same method (mostly NCBI refseq).
While PAV generally scales with divergence time, it is 2-4X more common in plants, especially those with a history of polyploids.
While PAV generally scales with divergence time, it is 2-4X more common in plants, especially those with a history of polyploids.

August 18, 2025 at 4:51 PM
We first looked at how divergence time correlates with gene PAV in pairs of plant and animal genomes that were annotated with the same method (mostly NCBI refseq).
While PAV generally scales with divergence time, it is 2-4X more common in plants, especially those with a history of polyploids.
While PAV generally scales with divergence time, it is 2-4X more common in plants, especially those with a history of polyploids.
Determining presence-absence variation (PAV) across reference genomes is a major goal of pangenome analysis. It turns out that A LOT of gene PAV is due to methodological artifacts.
We explore the causes of this in soybean and cotton datasets in our recent preprint: www.biorxiv.org/content/10.1...
We explore the causes of this in soybean and cotton datasets in our recent preprint: www.biorxiv.org/content/10.1...
August 18, 2025 at 4:51 PM
Determining presence-absence variation (PAV) across reference genomes is a major goal of pangenome analysis. It turns out that A LOT of gene PAV is due to methodological artifacts.
We explore the causes of this in soybean and cotton datasets in our recent preprint: www.biorxiv.org/content/10.1...
We explore the causes of this in soybean and cotton datasets in our recent preprint: www.biorxiv.org/content/10.1...
There are three major haplotypes that each harbor several large structural variants but few coding variants. While the evidence for single-marker associations was limited, these three typable haplotypes segregate major variation in dhurrin concentration and drought severity of source habitat


August 6, 2025 at 8:32 PM
There are three major haplotypes that each harbor several large structural variants but few coding variants. While the evidence for single-marker associations was limited, these three typable haplotypes segregate major variation in dhurrin concentration and drought severity of source habitat
Finally, we combined pangenome-informed haplotype classification and tests of drought adaptation by probing the biosynthetic gene cluster that produces dhurrin, a secondary metabolite known to enhance drought stress tolerance and resistance against chewing insect herbivory ...

August 6, 2025 at 8:32 PM
Finally, we combined pangenome-informed haplotype classification and tests of drought adaptation by probing the biosynthetic gene cluster that produces dhurrin, a secondary metabolite known to enhance drought stress tolerance and resistance against chewing insect herbivory ...
However, at a 110km scale, adjacent populations in the Northern (driest) part of the Sahel were far more different from each other than those from less drought-prone regions. This is in part caused by the admixture and potential local adaptation of different deeply diverged botanical types.


August 6, 2025 at 8:32 PM
However, at a 110km scale, adjacent populations in the Northern (driest) part of the Sahel were far more different from each other than those from less drought-prone regions. This is in part caused by the admixture and potential local adaptation of different deeply diverged botanical types.
We then explored variation in our resequencing panel to understand how deep divergence at SHATTERING1 and genome-wide diversity is shaped by climate, geography and migration.
At a continent-wide scale, gene flow was higher between populations in drought-prone habitats ...
At a continent-wide scale, gene flow was higher between populations in drought-prone habitats ...

August 6, 2025 at 8:32 PM
We then explored variation in our resequencing panel to understand how deep divergence at SHATTERING1 and genome-wide diversity is shaped by climate, geography and migration.
At a continent-wide scale, gene flow was higher between populations in drought-prone habitats ...
At a continent-wide scale, gene flow was higher between populations in drought-prone habitats ...
Here, we explored several genomic regions of interest and re-classified haplotype associations across our diversity panel using diagnostic short sequences. The approach discovered and genotyped a previously unknown exact >2kb duplication in the domestication locus SHATTERING1

August 6, 2025 at 8:32 PM
Here, we explored several genomic regions of interest and re-classified haplotype associations across our diversity panel using diagnostic short sequences. The approach discovered and genotyped a previously unknown exact >2kb duplication in the domestication locus SHATTERING1
We integrated these resources into a 'pangenome', a queryable resource w/ all available DNA sequences. We complimented the pangenome w/ RNA-seq evidenced protein-coding gene annotations of all 33 members. These resources capture the vast majority of gene presence-absence and DNA sequence variation.


August 6, 2025 at 8:32 PM
We integrated these resources into a 'pangenome', a queryable resource w/ all available DNA sequences. We complimented the pangenome w/ RNA-seq evidenced protein-coding gene annotations of all 33 members. These resources capture the vast majority of gene presence-absence and DNA sequence variation.
Sorghum bicolor is an incredibly diverse crop with 'grain' varieties that feed 500M people, 'sweet' types that produce sugary stalk juice, and fast-growing bioenergy feedstocks.
Here we built 33 reference genomes and short-read resequenced ~1900 varieties that span its genetic diversity.
Here we built 33 reference genomes and short-read resequenced ~1900 varieties that span its genetic diversity.

August 6, 2025 at 8:32 PM
Sorghum bicolor is an incredibly diverse crop with 'grain' varieties that feed 500M people, 'sweet' types that produce sugary stalk juice, and fast-growing bioenergy feedstocks.
Here we built 33 reference genomes and short-read resequenced ~1900 varieties that span its genetic diversity.
Here we built 33 reference genomes and short-read resequenced ~1900 varieties that span its genetic diversity.
@jrossibarra.bsky.social your kind of street art

May 26, 2025 at 4:29 PM
@jrossibarra.bsky.social your kind of street art
Biotechnology, including genome editing and marker assisted selection, offers a powerful method to accelerate gains along with RGS. To aide these efforts we built reference genomes for America and Chinese chestnut genotypes that founded The American Chestnut Foundations' (TACF) breeding program.

February 3, 2025 at 9:23 PM
Biotechnology, including genome editing and marker assisted selection, offers a powerful method to accelerate gains along with RGS. To aide these efforts we built reference genomes for America and Chinese chestnut genotypes that founded The American Chestnut Foundations' (TACF) breeding program.
This tradeoff necessitates an integrated approach to breeding: simultaneously selecting for resistance and high levels of C. dentata ancestry, a goal well suited for Recurrent Genomic Selection (‘RGS’). Optimized RGS may achieve resistant pops with >75% C. dentata ancestry in 1 or 2 generations


February 3, 2025 at 9:23 PM
This tradeoff necessitates an integrated approach to breeding: simultaneously selecting for resistance and high levels of C. dentata ancestry, a goal well suited for Recurrent Genomic Selection (‘RGS’). Optimized RGS may achieve resistant pops with >75% C. dentata ancestry in 1 or 2 generations
In contrast, we found that resistance is polygenic: in general, the more American ancestry, the more susceptible a tree is. However, there is a lot of variation and rare highly resistant genotypes exist with > 90% C. dentata.

February 3, 2025 at 9:23 PM
In contrast, we found that resistance is polygenic: in general, the more American ancestry, the more susceptible a tree is. However, there is a lot of variation and rare highly resistant genotypes exist with > 90% C. dentata.
Originally it was thought that few genetic loci controlled resistance, and backcrossing breeding would yield resistant trees with majority C. dentata ancestry.

February 3, 2025 at 9:23 PM
Originally it was thought that few genetic loci controlled resistance, and backcrossing breeding would yield resistant trees with majority C. dentata ancestry.
However, the line8 genome was far more difficult to assemble than a normal peanut: unexpected residual heterozygosity had to be collapsed & the chr9 introgression had to be split into alternative haplotypes, producing the first in situ resolved sequence of the Chr9 A. cardenasii introgression.

November 12, 2024 at 11:05 PM
However, the line8 genome was far more difficult to assemble than a normal peanut: unexpected residual heterozygosity had to be collapsed & the chr9 introgression had to be split into alternative haplotypes, producing the first in situ resolved sequence of the Chr9 A. cardenasii introgression.
Complicating genome assembly further, several peanut genomes commonly undergo homeologous exchange where 'tetrasomic' regions on the ends of several chromosomes recombine and are nearly identical between the subgenomes.

November 12, 2024 at 11:05 PM
Complicating genome assembly further, several peanut genomes commonly undergo homeologous exchange where 'tetrasomic' regions on the ends of several chromosomes recombine and are nearly identical between the subgenomes.
Peanut already has a difficult genome to assemble: it is pretty big (2.6Mb), tetraploid, and highly repetitive. Ty3 repeats make up > 50% of the entire genome sequence!

November 12, 2024 at 11:05 PM
Peanut already has a difficult genome to assemble: it is pretty big (2.6Mb), tetraploid, and highly repetitive. Ty3 repeats make up > 50% of the entire genome sequence!
Mid-pandemic, Phat Dang at the USDA and Charles Chen at the Auburn University peanut breeding program found that the 8th line from a population bred for drought tolerance yielded far better than its siblings under a rainout shelter. We creatively called this cultivar 'line8'.

November 12, 2024 at 11:05 PM
Mid-pandemic, Phat Dang at the USDA and Charles Chen at the Auburn University peanut breeding program found that the 8th line from a population bred for drought tolerance yielded far better than its siblings under a rainout shelter. We creatively called this cultivar 'line8'.
We combined many data sources including @PacBio HiFi, HiC, @Illumina sequencing of single chromosome sorts, and optical & genetic maps. This produced a genome that presents the full set of biological sequence variation in the R570 hybrid cultivar.

March 29, 2024 at 5:19 PM
We combined many data sources including @PacBio HiFi, HiC, @Illumina sequencing of single chromosome sorts, and optical & genetic maps. This produced a genome that presents the full set of biological sequence variation in the R570 hybrid cultivar.