



Josipa Lipovac
@jlipovac.bsky.social
PhD student at FER, University of Zagreb | Bioinformatics | Metagenome analysis | Genome assembly
Congrats Martin! 🥳🥳
November 7, 2025 at 9:19 AM
Congrats Martin! 🥳🥳
Thanks Roland! It’s good to mention here that Hairsplitter is also part of it 😄
May 16, 2025 at 6:01 PM
Thanks Roland! It’s good to mention here that Hairsplitter is also part of it 😄
Work with @msikic.bsky.social, @rvicedomini.bsky.social, Kresimir Krizanovic
MADRe is open-source, modular, and ready to use.
Check it out:
🔗 github.com/lbcb-sci/MADRe
9/9
MADRe is open-source, modular, and ready to use.
Check it out:
🔗 github.com/lbcb-sci/MADRe
9/9

GitHub - lbcb-sci/MADRe: Strain-level metagenomic classification with Metagenome Assembly driven Database Reduction approach
Strain-level metagenomic classification with Metagenome Assembly driven Database Reduction approach - lbcb-sci/MADRe
github.com
May 16, 2025 at 8:37 AM
Work with @msikic.bsky.social, @rvicedomini.bsky.social, Kresimir Krizanovic
MADRe is open-source, modular, and ready to use.
Check it out:
🔗 github.com/lbcb-sci/MADRe
9/9
MADRe is open-source, modular, and ready to use.
Check it out:
🔗 github.com/lbcb-sci/MADRe
9/9
A key feature of MADRe is its focus on organisms with sufficient abundance to be assembled.
While low-abundance strains may be underrepresented, this trade-off significantly reduces false-positive identifications, a common issue in strain-level metagenomics.
8/9
While low-abundance strains may be underrepresented, this trade-off significantly reduces false-positive identifications, a common issue in strain-level metagenomics.
8/9
May 16, 2025 at 8:37 AM
A key feature of MADRe is its focus on organisms with sufficient abundance to be assembled.
While low-abundance strains may be underrepresented, this trade-off significantly reduces false-positive identifications, a common issue in strain-level metagenomics.
8/9
While low-abundance strains may be underrepresented, this trade-off significantly reduces false-positive identifications, a common issue in strain-level metagenomics.
8/9
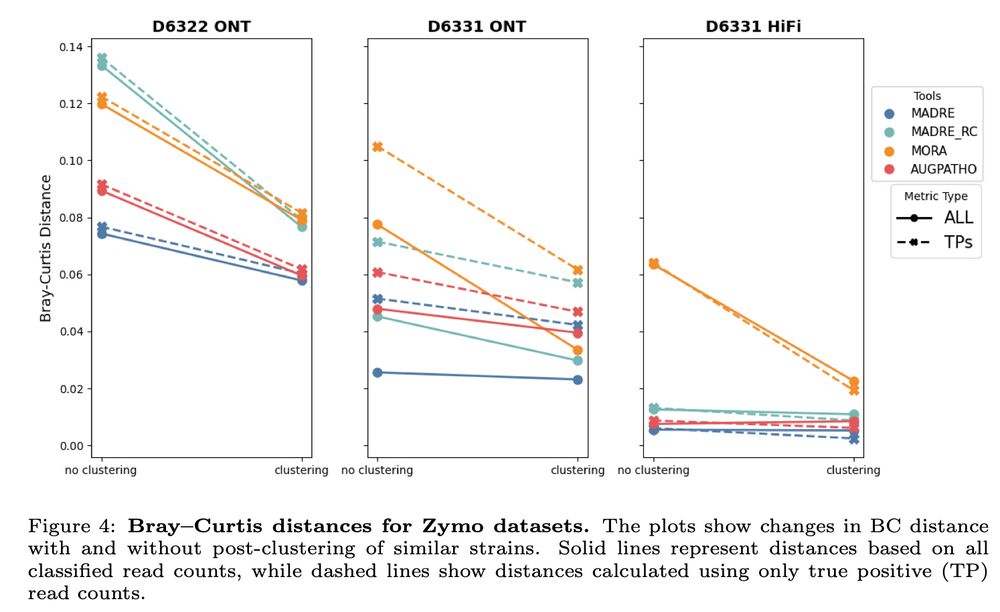
We evaluated MADRe on both real and simulated datasets and observed:
✅ Comparable or improved accuracy over existing tools
✅ Clearer and more realistic abundance profiles
✅ Substantial reductions in runtime and memory usage
7/9
✅ Comparable or improved accuracy over existing tools
✅ Clearer and more realistic abundance profiles
✅ Substantial reductions in runtime and memory usage
7/9

May 16, 2025 at 8:37 AM
We evaluated MADRe on both real and simulated datasets and observed:
✅ Comparable or improved accuracy over existing tools
✅ Clearer and more realistic abundance profiles
✅ Substantial reductions in runtime and memory usage
7/9
✅ Comparable or improved accuracy over existing tools
✅ Clearer and more realistic abundance profiles
✅ Substantial reductions in runtime and memory usage
7/9
While assembly is often considered computationally expensive, we demonstrate that MADRe, by combining assembly with contig-level mapping, is more efficient than directly mapping large volumes of reads to a full reference database. 6/9

May 16, 2025 at 8:37 AM
While assembly is often considered computationally expensive, we demonstrate that MADRe, by combining assembly with contig-level mapping, is more efficient than directly mapping large volumes of reads to a full reference database. 6/9
To complete the pipeline, MADRe maps reads to the reduced reference database and applies a second round of probabilistic reassignment.
This enhances classification sensitivity and filters false-positive identifications, enabling precise strain-level profiling. 5/9
This enhances classification sensitivity and filters false-positive identifications, enabling precise strain-level profiling. 5/9

May 16, 2025 at 8:37 AM
To complete the pipeline, MADRe maps reads to the reduced reference database and applies a second round of probabilistic reassignment.
This enhances classification sensitivity and filters false-positive identifications, enabling precise strain-level profiling. 5/9
This enhances classification sensitivity and filters false-positive identifications, enabling precise strain-level profiling. 5/9
This reduction identifies candidate genomes present in the sample.
However, this step alone does not eliminate all false positives and does not provide accurate abundance estimates. 4/9
However, this step alone does not eliminate all false positives and does not provide accurate abundance estimates. 4/9
May 16, 2025 at 8:37 AM
This reduction identifies candidate genomes present in the sample.
However, this step alone does not eliminate all false positives and does not provide accurate abundance estimates. 4/9
However, this step alone does not eliminate all false positives and does not provide accurate abundance estimates. 4/9
MADRe begins by assembling the metagenomic sample and mapping the resulting contigs - often representing collapsed strains - to a (large) reference database.
Using EM-based read reassignment and info about strain collapses, we construct reduced database. 3/9
Using EM-based read reassignment and info about strain collapses, we construct reduced database. 3/9
May 16, 2025 at 8:37 AM
MADRe begins by assembling the metagenomic sample and mapping the resulting contigs - often representing collapsed strains - to a (large) reference database.
Using EM-based read reassignment and info about strain collapses, we construct reduced database. 3/9
Using EM-based read reassignment and info about strain collapses, we construct reduced database. 3/9
Strain-level classification requires large reference databases, especially when there is no prior knowledge about sample composition.
However, mapping reads to such large databases is computationally expensive and often impractical at scale. 2/9
However, mapping reads to such large databases is computationally expensive and often impractical at scale. 2/9
May 16, 2025 at 8:37 AM
Strain-level classification requires large reference databases, especially when there is no prior knowledge about sample composition.
However, mapping reads to such large databases is computationally expensive and often impractical at scale. 2/9
However, mapping reads to such large databases is computationally expensive and often impractical at scale. 2/9