



David Ryan Koes
@dkoes.compstruct.org
Removing barriers to computational drug discovery one bit at a time. Associate Professor in Computational and Systems Biology at the University of Pittsburgh.
https://bits.csb.pitt.edu/
https://bits.csb.pitt.edu/
Was proud and honored to hood Dr. Drew McNutt at the Pitt School of Medicine Diploma Ceremony. Here we are rocking both the blue and gold and tartan colors representing our joint Pitt-CMU CompBio PhD program. Congratulations to Drew and the other @cmupittcompbio.bsky.social graduates!


May 19, 2025 at 1:55 PM
Was proud and honored to hood Dr. Drew McNutt at the Pitt School of Medicine Diploma Ceremony. Here we are rocking both the blue and gold and tartan colors representing our joint Pitt-CMU CompBio PhD program. Congratulations to Drew and the other @cmupittcompbio.bsky.social graduates!
Interested in generative modeling and pharmacophores search for SBDD? Check out our talks at #ACSSpring2025

March 25, 2025 at 3:15 PM
Interested in generative modeling and pharmacophores search for SBDD? Check out our talks at #ACSSpring2025


March 7, 2025 at 4:56 PM
Dang - forget BlueSky makes transparent backgrounds black. There's a hidden message.

December 15, 2024 at 6:59 PM
Dang - forget BlueSky makes transparent backgrounds black. There's a hidden message.
I think this is less important for the train set compared to getting a pristine, possibly manually curated test. A data loader that downsampled duplicated ligands would go a long way to smoothing out problematic systems.
December 15, 2024 at 6:52 PM
I think this is less important for the train set compared to getting a pristine, possibly manually curated test. A data loader that downsampled duplicated ligands would go a long way to smoothing out problematic systems.
It turns out that the picomolar ligand BMP (www.rcsb.org/ligand/BMP) is present 30 times in the test set. It isn't clear where this affinity was sourced from and the structures include mutant proteins that presumably should have different binding affinities (but they are all labeled the same).

December 15, 2024 at 6:52 PM
It turns out that the picomolar ligand BMP (www.rcsb.org/ligand/BMP) is present 30 times in the test set. It isn't clear where this affinity was sourced from and the structures include mutant proteins that presumably should have different binding affinities (but they are all labeled the same).
Now onto the less good... When @alutsky.bsky.social trained a binding affinity prediction model on PLINDER, he got a negative correlation, which was surprising looking at the predictions (note only 182 test set systems having an affinity annotation).

December 15, 2024 at 6:52 PM
Now onto the less good... When @alutsky.bsky.social trained a binding affinity prediction model on PLINDER, he got a negative correlation, which was surprising looking at the predictions (note only 182 test set systems having an affinity annotation).
Using GraphPocket descriptors (developed in @vratin.bsky.social's MS thesis) there is some separation, but there remains a significant amount of overlap.

December 15, 2024 at 6:52 PM
Using GraphPocket descriptors (developed in @vratin.bsky.social's MS thesis) there is some separation, but there remains a significant amount of overlap.
Interestingly, using distances between learned descriptors gets different results. Using DeeplyTough descriptors the test set is as close to the training set as to the removed set.

December 15, 2024 at 6:52 PM
Interestingly, using distances between learned descriptors gets different results. Using DeeplyTough descriptors the test set is as close to the training set as to the removed set.
Here are persistent homology descriptors calculated by @alutsky.bsky.social (from his MS thesis). This descriptor space has a different scale.

December 15, 2024 at 6:52 PM
Here are persistent homology descriptors calculated by @alutsky.bsky.social (from his MS thesis). This descriptor space has a different scale.
Using geometric shape descriptors of the pocket, the test set is separated from training set. Using Zernike descriptors calculated by @katztyler.bsky.social we see that the closest system to the test examples have been removed from training and the train set is not close to the test set.

December 15, 2024 at 6:52 PM
Using geometric shape descriptors of the pocket, the test set is separated from training set. Using Zernike descriptors calculated by @katztyler.bsky.social we see that the closest system to the test examples have been removed from training and the train set is not close to the test set.

December 15, 2024 at 6:21 PM
My wife gave this to me today. Good news - it is empty!

December 6, 2024 at 12:20 PM
My wife gave this to me today. Good news - it is empty!
You lost me with the use of the singular…

November 29, 2024 at 3:03 AM
You lost me with the use of the singular…
Unlike previous approaches, SPRINT uses attention to learn from embeddings, resulting in some level of interpretability. It turns out the attention mask is sparse with some preference for binding site residues but more for less conserved residues.

November 26, 2024 at 1:28 PM
Unlike previous approaches, SPRINT uses attention to learn from embeddings, resulting in some level of interpretability. It turns out the attention mask is sparse with some preference for binding site residues but more for less conserved residues.
Results on LIT-PCBA (an admittedly somewhat problematic benchmark) are quite good, although I personally don't see this replacing more traditional techniques so much as supplementing them. We're looking into using it to initialize "deep docking" runs.

November 26, 2024 at 1:28 PM
Results on LIT-PCBA (an admittedly somewhat problematic benchmark) are quite good, although I personally don't see this replacing more traditional techniques so much as supplementing them. We're looking into using it to initialize "deep docking" runs.
We're looking forward to presenting this @workshopmlsb.bsky.social . This work was conceived and led by a group CPCB (@cmupittcompbio.bsky.social) students and learns a co-embedding of proteins and ligands to support ultra fast virtual screening. arxiv.org/pdf/2411.15418


November 26, 2024 at 1:28 PM
We're looking forward to presenting this @workshopmlsb.bsky.social . This work was conceived and led by a group CPCB (@cmupittcompbio.bsky.social) students and learns a co-embedding of proteins and ligands to support ultra fast virtual screening. arxiv.org/pdf/2411.15418
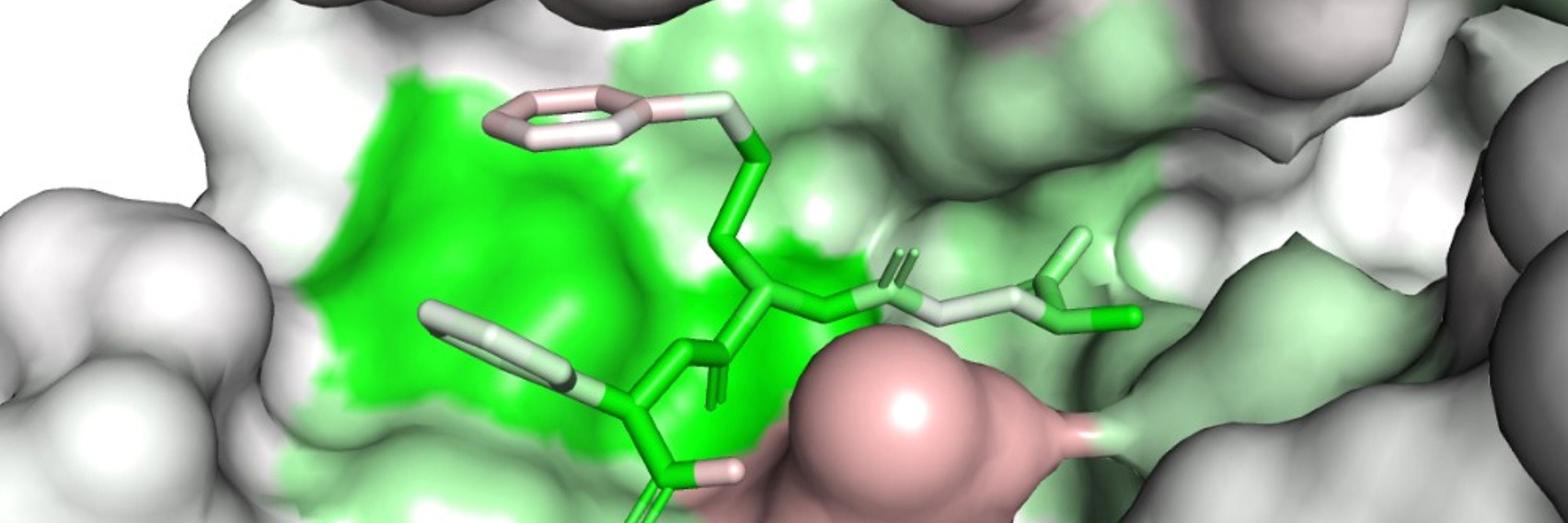
This is why I've been looking at profilin - there are ALS linked mutations that open up a destabilizing pocket (yellow). Boltz-1 gets the pocket right (unsurprising - it is in the training set), but not a single ligand gets docked there. Maybe this means it is "undruggable", but I doubt it.

November 25, 2024 at 6:45 PM
This is why I've been looking at profilin - there are ALS linked mutations that open up a destabilizing pocket (yellow). Boltz-1 gets the pocket right (unsurprising - it is in the training set), but not a single ligand gets docked there. Maybe this means it is "undruggable", but I doubt it.
Here is how Boltz-1 (green), DynamicBind (magenta), and GNINA (blue) dock a collection of random molecules. GNINA, using a classical sampling algorithm (MCMC) hits all concave regions while the ML samplers have distinct preferences. Boltz is the most likely to induce a fit.
November 22, 2024 at 6:27 PM
Here is how Boltz-1 (green), DynamicBind (magenta), and GNINA (blue) dock a collection of random molecules. GNINA, using a classical sampling algorithm (MCMC) hits all concave regions while the ML samplers have distinct preferences. Boltz is the most likely to induce a fit.
Here's how the ligands dock. Clearly there are some preferred binding sites.

November 21, 2024 at 10:06 PM
Here's how the ligands dock. Clearly there are some preferred binding sites.
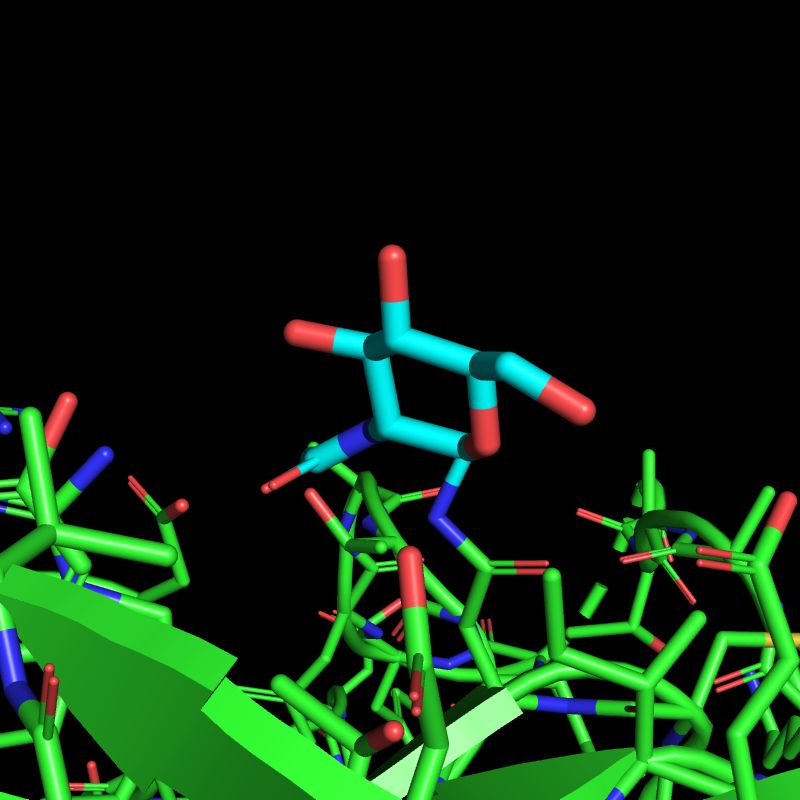
Some fun with Boltz-1 (www.biorxiv.org/content/10.1...). I generated 1000 samples profilin (green) and compare them to the NMR structures (cyan). Samples were generated by docking 1000 random ligands. NMR structures show more conformational diversity.
November 21, 2024 at 9:40 PM
Some fun with Boltz-1 (www.biorxiv.org/content/10.1...). I generated 1000 samples profilin (green) and compare them to the NMR structures (cyan). Samples were generated by docking 1000 random ligands. NMR structures show more conformational diversity.
Excellent work from Ian Dunn
x.com/ian_dunn_/st...
x.com/ian_dunn_/st...

May 1, 2024 at 1:06 PM
Excellent work from Ian Dunn
x.com/ian_dunn_/st...
x.com/ian_dunn_/st...