




Alex Geary
@alextremophile.bsky.social
Postdoctoral Bioinformaticial in the Computational Rare Disease Genomics group (Nicky Whiffin). univ. Oxford 🧬💻
Loves Evolution, regulation, cheese and cats. She/Her
Loves Evolution, regulation, cheese and cats. She/Her
And a special shout out to the absolutely awesome senior author on this - @ruebenadawes.bsky.social who was a total delight to work with from start to finish!

a cartoon character says you are my hero
ALT: a cartoon character says you are my hero
media.tenor.com
August 29, 2025 at 8:57 AM
And a special shout out to the absolutely awesome senior author on this - @ruebenadawes.bsky.social who was a total delight to work with from start to finish!
I would also like to thank all my co-authors for the time, effort and input that has made this a super little bit of work! I'm looking at you @francois-leco.bsky.social Suzi Walker and @nickywhiffin.bsky.social 👀
August 29, 2025 at 8:57 AM
I would also like to thank all my co-authors for the time, effort and input that has made this a super little bit of work! I'm looking at you @francois-leco.bsky.social Suzi Walker and @nickywhiffin.bsky.social 👀
We would like to thank everyone who has contributed to this work, but most of all the team at Genomics England, all the participants and their families, without whom all this would not have been possible.

a man with long hair and a beard is smiling and saying thank you .
ALT: a man with long hair and a beard is smiling and saying thank you .
media.tenor.com
August 29, 2025 at 8:57 AM
We would like to thank everyone who has contributed to this work, but most of all the team at Genomics England, all the participants and their families, without whom all this would not have been possible.
We therefore developed a command-line tool, ‘SpliceAI-splint’, to identify the subset of variants for which the precomputed scores may have decreased accuracy, to allow reannotation of a smaller overall variant set. github.com/Computationa...
August 29, 2025 at 8:57 AM
We therefore developed a command-line tool, ‘SpliceAI-splint’, to identify the subset of variants for which the precomputed scores may have decreased accuracy, to allow reannotation of a smaller overall variant set. github.com/Computationa...
These findings have real implications for the continued use of precomputed scores. However, despite these issues, precomputed scores still have substantial utility in limiting the computational resources required to identify predicted splice-altering variants.
August 29, 2025 at 8:57 AM
These findings have real implications for the continued use of precomputed scores. However, despite these issues, precomputed scores still have substantial utility in limiting the computational resources required to identify predicted splice-altering variants.
By correcting these issues we found an increase of 18.2% predicted splice-altering variants compared with variants identified using precomputed scores alone. When assessed using diagnostic variants, updated SpliceAI scores resulted in a diagnostic increase of 11.7%.

August 29, 2025 at 8:57 AM
By correcting these issues we found an increase of 18.2% predicted splice-altering variants compared with variants identified using precomputed scores alone. When assessed using diagnostic variants, updated SpliceAI scores resulted in a diagnostic increase of 11.7%.
To determine the potential impact of these issues we annotated variants in a subset of participants recruited with neurodevelopmental disorders in the @genomicsengland.bsky.social National Genomics Research Library, using both precomputed scores, and SpliceAI with updated parameters.

Homepage
Genomics England analyses sequenced genomes for the NHS and then equips researchers to use data to help find the cause of disease.
www.genomicsengland.co.uk
August 29, 2025 at 8:57 AM
To determine the potential impact of these issues we annotated variants in a subset of participants recruited with neurodevelopmental disorders in the @genomicsengland.bsky.social National Genomics Research Library, using both precomputed scores, and SpliceAI with updated parameters.
Precomputed scores were calculated using a distance parameter of 50nt, however the suggested threshold has increased to 500nt. They can’t handle larger insertions and deletions. And there are errors within the file itself, likely arising from conversion of the original annotations to GRCh38.
August 29, 2025 at 8:57 AM
Precomputed scores were calculated using a distance parameter of 50nt, however the suggested threshold has increased to 500nt. They can’t handle larger insertions and deletions. And there are errors within the file itself, likely arising from conversion of the original annotations to GRCh38.
Unfortunately these scores have limitations that can reduce their accuracy in certain contexts: For example changes in transcript annotations, altered transcript boundaries, absence of annotated genes, and changes to exon composition can mean that scores are missed or altered.

August 29, 2025 at 8:57 AM
Unfortunately these scores have limitations that can reduce their accuracy in certain contexts: For example changes in transcript annotations, altered transcript boundaries, absence of annotated genes, and changes to exon composition can mean that scores are missed or altered.
These scores also underlie spliceAI annotations in many variant effect predictors such as the @ensembl.org VEP, forming the basis for annotation in many diagnostic pipelines.
Ensembl Variant Effect Predictor (VEP)
www.ensembl.org
August 29, 2025 at 8:57 AM
These scores also underlie spliceAI annotations in many variant effect predictors such as the @ensembl.org VEP, forming the basis for annotation in many diagnostic pipelines.
SpliceAI is an invaluable tool to identify splice-altering variants. The provision of precomputed scores for all theoretically possible SNVs, 1 base insertions, and 1-4 base deletions has extended this further, massively reducing the time and cost associated with running spliceAI directly.
SpliceAI Lookup
spliceailookup.broadinstitute.org
August 29, 2025 at 8:57 AM
SpliceAI is an invaluable tool to identify splice-altering variants. The provision of precomputed scores for all theoretically possible SNVs, 1 base insertions, and 1-4 base deletions has extended this further, massively reducing the time and cost associated with running spliceAI directly.
Reposted by Alex Geary

I learned so much from this work and I hope that and discoveries like this can make a real difference to the lives of people living with #RareConditions. Please do share! 😊
www.medrxiv.org/content/10.1...
www.medrxiv.org/content/10.1...

Biallelic variants in the non-coding RNA gene RNU4-2 cause a recessive neurodevelopmental syndrome with distinct white matter changes
Genetic variants in RNU4-2, which encodes U4, a key non-coding small nuclear RNA (snRNA) component of the major spliceosome, were recently shown to cause a prevalent neurodevelopmental disorder (NDD) ...
www.medrxiv.org
August 18, 2025 at 11:23 AM
I learned so much from this work and I hope that and discoveries like this can make a real difference to the lives of people living with #RareConditions. Please do share! 😊
www.medrxiv.org/content/10.1...
www.medrxiv.org/content/10.1...
Reposted by Alex Geary
The headlines?
1) Variants on both copies of #RNU4-2 cause a recessive neurodevelopmental disorder with prominent speech delay
2) One of the hallmarks is distinct white matter changes on MRI
3) It is clinically and genetically distinct from #ReNU syndrome
www.medrxiv.org/content/10.1...
1) Variants on both copies of #RNU4-2 cause a recessive neurodevelopmental disorder with prominent speech delay
2) One of the hallmarks is distinct white matter changes on MRI
3) It is clinically and genetically distinct from #ReNU syndrome
www.medrxiv.org/content/10.1...
Biallelic variants in the non-coding RNA gene RNU4-2 cause a recessive neurodevelopmental syndrome with distinct white matter changes
Genetic variants in RNU4-2, which encodes U4, a key non-coding small nuclear RNA (snRNA) component of the major spliceosome, were recently shown to cause a prevalent neurodevelopmental disorder (NDD) ...
www.medrxiv.org
August 18, 2025 at 11:23 AM
The headlines?
1) Variants on both copies of #RNU4-2 cause a recessive neurodevelopmental disorder with prominent speech delay
2) One of the hallmarks is distinct white matter changes on MRI
3) It is clinically and genetically distinct from #ReNU syndrome
www.medrxiv.org/content/10.1...
1) Variants on both copies of #RNU4-2 cause a recessive neurodevelopmental disorder with prominent speech delay
2) One of the hallmarks is distinct white matter changes on MRI
3) It is clinically and genetically distinct from #ReNU syndrome
www.medrxiv.org/content/10.1...
Reposted by Alex Geary

3/
The review outlines how UTR variants cause disease, such as:
- Create or remove upstream AUGs (uAUGs)
- Alter splicing
- Alter polyadenylation
- Interfere with miRNA or protein binding
The review outlines how UTR variants cause disease, such as:
- Create or remove upstream AUGs (uAUGs)
- Alter splicing
- Alter polyadenylation
- Interfere with miRNA or protein binding
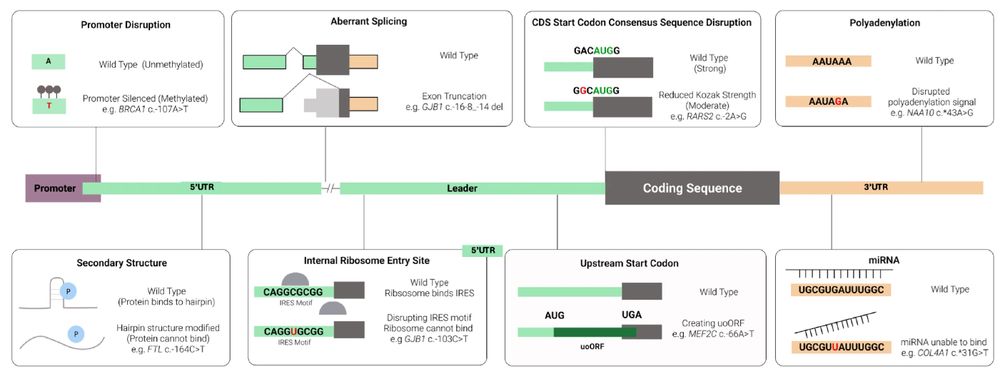
July 4, 2025 at 8:08 AM
3/
The review outlines how UTR variants cause disease, such as:
- Create or remove upstream AUGs (uAUGs)
- Alter splicing
- Alter polyadenylation
- Interfere with miRNA or protein binding
The review outlines how UTR variants cause disease, such as:
- Create or remove upstream AUGs (uAUGs)
- Alter splicing
- Alter polyadenylation
- Interfere with miRNA or protein binding
Finally, and most importantly - this would not have been possible without the support, and contributions made by all the participants and families enrolled in @genomicsengland.bsky.social, their clinical teams, and the staff at Genomics England - to whom we are eternally grateful.

a white puppy is laying on the ground with the words `` thank you thank you thank you thank you '' written above it .
ALT: a white puppy is laying on the ground with the words `` thank you thank you thank you thank you '' written above it .
media.tenor.com
April 14, 2025 at 5:43 PM
Finally, and most importantly - this would not have been possible without the support, and contributions made by all the participants and families enrolled in @genomicsengland.bsky.social, their clinical teams, and the staff at Genomics England - to whom we are eternally grateful.
I would like to give my heartfelt thanks to everyone who has contributed to this work, in particular the incredible @nickywhiffin.bsky.social, Alex Blakes, @drjennylord.bsky.social , Sid Banka and Scott Findlay, who have worked very hard to get this out.
April 14, 2025 at 5:43 PM
I would like to give my heartfelt thanks to everyone who has contributed to this work, in particular the incredible @nickywhiffin.bsky.social, Alex Blakes, @drjennylord.bsky.social , Sid Banka and Scott Findlay, who have worked very hard to get this out.
The take 🏡 :
Variants that occur outside of protein-coding regions represent a modest but appreciable contribution to rare disease, and should be routinely incorporated into diagnostic pipelines.
(We present a systematic framework for doing this!)
Variants that occur outside of protein-coding regions represent a modest but appreciable contribution to rare disease, and should be routinely incorporated into diagnostic pipelines.
(We present a systematic framework for doing this!)
April 14, 2025 at 5:43 PM
The take 🏡 :
Variants that occur outside of protein-coding regions represent a modest but appreciable contribution to rare disease, and should be routinely incorporated into diagnostic pipelines.
(We present a systematic framework for doing this!)
Variants that occur outside of protein-coding regions represent a modest but appreciable contribution to rare disease, and should be routinely incorporated into diagnostic pipelines.
(We present a systematic framework for doing this!)
Looking at the burden that these variants may represent in rare disease we did see an appreciable increase in variants with the potential to disrupt regulation in cases vs controls, though this did not meet the threshold for significance.
link.springer.com/article/10.1...
link.springer.com/article/10.1...

April 14, 2025 at 5:43 PM
Looking at the burden that these variants may represent in rare disease we did see an appreciable increase in variants with the potential to disrupt regulation in cases vs controls, though this did not meet the threshold for significance.
link.springer.com/article/10.1...
link.springer.com/article/10.1...