




Alan Amin
@alannawzadamin.bsky.social
Faculty fellow at NYU working with @andrewgwils.bsky.social. Statistics & machine learning for proteins, RNA, DNA.
Prev: @jura.bsky.social, PhD with Debora Marks
Website: alannawzadamin.github.io
Prev: @jura.bsky.social, PhD with Debora Marks
Website: alannawzadamin.github.io
Many thanks for the award for this work at AI4NA workshop at ICLR!
More experiments and details of our linear algebra in the paper! Come say hi at ICML! 7/7
Paper: arxiv.org/abs/2506.19598
Code: github.com/AlanNawzadAm...
More experiments and details of our linear algebra in the paper! Come say hi at ICML! 7/7
Paper: arxiv.org/abs/2506.19598
Code: github.com/AlanNawzadAm...

June 25, 2025 at 2:03 PM
Many thanks for the award for this work at AI4NA workshop at ICLR!
More experiments and details of our linear algebra in the paper! Come say hi at ICML! 7/7
Paper: arxiv.org/abs/2506.19598
Code: github.com/AlanNawzadAm...
More experiments and details of our linear algebra in the paper! Come say hi at ICML! 7/7
Paper: arxiv.org/abs/2506.19598
Code: github.com/AlanNawzadAm...
Ablations on model size and number of features show that larger models trained on more features make more accurate predictions with no evidence of plateauing! This suggests further improvements by training across many phenotypes, or across populations in future work! 6/7

June 25, 2025 at 2:03 PM
Ablations on model size and number of features show that larger models trained on more features make more accurate predictions with no evidence of plateauing! This suggests further improvements by training across many phenotypes, or across populations in future work! 6/7
We can now train flexible models on many features. DeepWAS models predict significant enrichment of effect in conserved regions, accessible chromatin, and TF binding sites! And, as shown above, they make better phenotype predictions in practice! 5/7

June 25, 2025 at 2:03 PM
We can now train flexible models on many features. DeepWAS models predict significant enrichment of effect in conserved regions, accessible chromatin, and TF binding sites! And, as shown above, they make better phenotype predictions in practice! 5/7
Our idea is to rearrange the linear algebra problem to counter-intuitively increase matrix size, but make it "better conditioned". Iterative algorithms (like CG) converge on huge matrices quickly! By moving to GPU we achieved another order of magnitude speed up! 4/7

June 25, 2025 at 2:03 PM
Our idea is to rearrange the linear algebra problem to counter-intuitively increase matrix size, but make it "better conditioned". Iterative algorithms (like CG) converge on huge matrices quickly! By moving to GPU we achieved another order of magnitude speed up! 4/7
But to train on the full likelihood, we must solve big linear algebra problems because variants in the genome are correlated. A naive method would take O(M³) – intractable for many variants! By reformulating the problem, we reduce this to roughly O(M²), enabling large models. 3/7

June 25, 2025 at 2:03 PM
But to train on the full likelihood, we must solve big linear algebra problems because variants in the genome are correlated. A naive method would take O(M³) – intractable for many variants! By reformulating the problem, we reduce this to roughly O(M²), enabling large models. 3/7
Many previous methods used the computationally light “LDSR objective” and saw no benefit from larger models – maybe deep learning isn’t useful here? No! Using the full likelihood, DeepWAS unlocks the potential of deep priors, improving phenotype prediction on UK Biobank! 2/7

June 25, 2025 at 2:03 PM
Many previous methods used the computationally light “LDSR objective” and saw no benefit from larger models – maybe deep learning isn’t useful here? No! Using the full likelihood, DeepWAS unlocks the potential of deep priors, improving phenotype prediction on UK Biobank! 2/7
We can make population genetics studies more powerful by building priors of variant effect size from features like binding. But we’ve been stuck on linear models! We introduce DeepWAS to learn deep priors on millions of variants! #ICML2025 Andres Potapczynski, @andrewgwils.bsky.social 1/7

June 25, 2025 at 2:03 PM
We can make population genetics studies more powerful by building priors of variant effect size from features like binding. But we’ve been stuck on linear models! We introduce DeepWAS to learn deep priors on millions of variants! #ICML2025 Andres Potapczynski, @andrewgwils.bsky.social 1/7
When applying SCUD to gradual processes like Gaussian (images) and BLOSUM (proteins), we combine masking's scheduling advantage with domain-specific inductive biases, outperforming both masking and classical diffusion! 6/7

June 16, 2025 at 2:20 PM
When applying SCUD to gradual processes like Gaussian (images) and BLOSUM (proteins), we combine masking's scheduling advantage with domain-specific inductive biases, outperforming both masking and classical diffusion! 6/7
We show that controlling the amount of information about transition times in SCUD interpolates between uniform noise and masking, clearly illustrating why masking has superior performance. But SCUD also applies to other forward processes!

June 16, 2025 at 2:20 PM
We show that controlling the amount of information about transition times in SCUD interpolates between uniform noise and masking, clearly illustrating why masking has superior performance. But SCUD also applies to other forward processes!
That’s exactly what we do to build schedule-conditioned diffusion (SCUD) models! After some math, training a SCUD model is like training a classical model except time is replaced with the number of transitions at each position, a soft version of how “masked” each position is! 4/7

June 16, 2025 at 2:20 PM
That’s exactly what we do to build schedule-conditioned diffusion (SCUD) models! After some math, training a SCUD model is like training a classical model except time is replaced with the number of transitions at each position, a soft version of how “masked” each position is! 4/7
But in practice, SOTA diffusion models have detectable errors in transition times! The exception is masking, which is typically parameterized to bake-in the known distribution of “when”. Why don’t we represent this knowledge in other discrete diffusion models? 3/7

June 16, 2025 at 2:20 PM
But in practice, SOTA diffusion models have detectable errors in transition times! The exception is masking, which is typically parameterized to bake-in the known distribution of “when”. Why don’t we represent this knowledge in other discrete diffusion models? 3/7
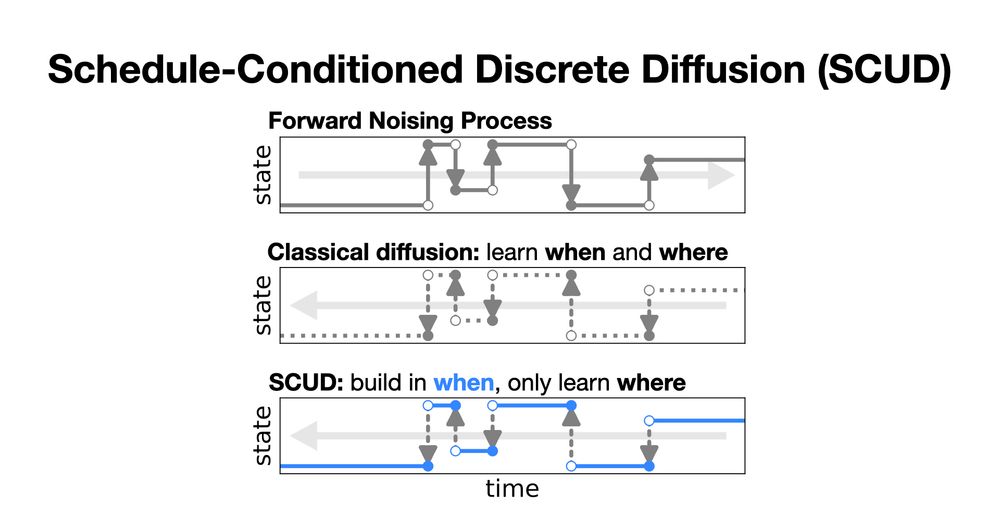
In discrete space, the forward noising process involves jump transitions between states. Reversing these paths involves learning when and where to transition. Often the “when” is known in closed form a priori, so it should be easy to learn… 2/7

June 16, 2025 at 2:20 PM
In discrete space, the forward noising process involves jump transitions between states. Reversing these paths involves learning when and where to transition. Often the “when” is known in closed form a priori, so it should be easy to learn… 2/7
There are many domain-specific noise processes for discrete diffusion, but masking dominates! Why? We show masking exploits a key property of discrete diffusion, which we use to unlock the potential of those structured processes and beat masking! w/ Nate Gruver and @andrewgwils.bsky.social 1/7
June 16, 2025 at 2:20 PM
There are many domain-specific noise processes for discrete diffusion, but masking dominates! Why? We show masking exploits a key property of discrete diffusion, which we use to unlock the potential of those structured processes and beat masking! w/ Nate Gruver and @andrewgwils.bsky.social 1/7
Finally, we validated CloneBO in vitro! We did one round of designs and tested them in the lab, comparing against the next best method. We see that CloneBO’s designs improve stability and significantly beat LaMBO-Ab in binding. 6/7

December 17, 2024 at 4:01 PM
Finally, we validated CloneBO in vitro! We did one round of designs and tested them in the lab, comparing against the next best method. We see that CloneBO’s designs improve stability and significantly beat LaMBO-Ab in binding. 6/7
To use our prior to optimize an antibody, we now need to generate clonal families that match measurements in the lab – bad mutations should be unlikely and good mutations likely. We developed a twisted sequential Monte Carlo approach to efficiently sample from this posterior. 5/7

December 17, 2024 at 4:01 PM
To use our prior to optimize an antibody, we now need to generate clonal families that match measurements in the lab – bad mutations should be unlikely and good mutations likely. We developed a twisted sequential Monte Carlo approach to efficiently sample from this posterior. 5/7
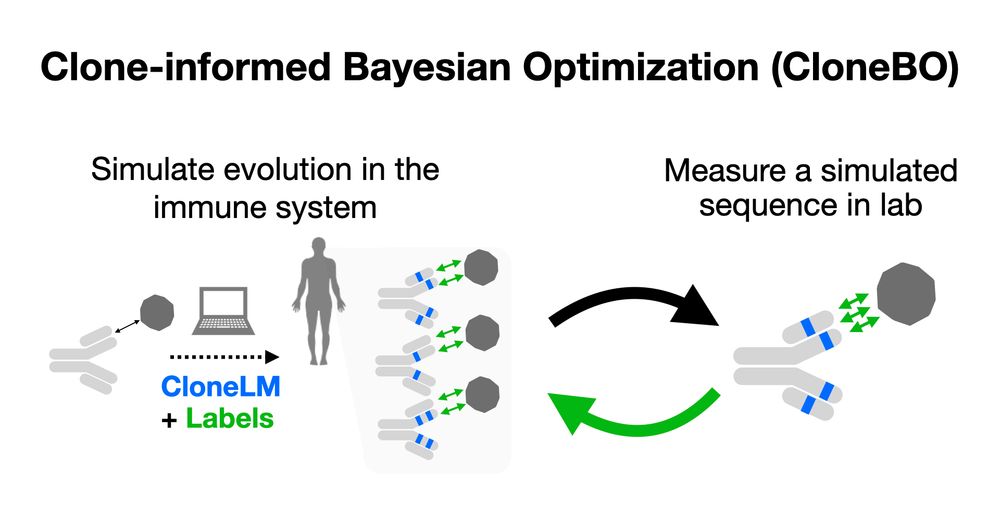
We train a transformer model to generate entire clonal families – CloneLM. Prompting with a single sequence, CloneLM samples realistic clonal families. These samples represent a prior on possible evolutionary trajectories in the immune system. 4/7

December 17, 2024 at 4:01 PM
We train a transformer model to generate entire clonal families – CloneLM. Prompting with a single sequence, CloneLM samples realistic clonal families. These samples represent a prior on possible evolutionary trajectories in the immune system. 4/7
Our bodies make antibodies by evolving specific portions of their sequences to bind their target strongly and stably, resulting in a set of related sequences known as a clonal family. We leverage modern software and data to build a dataset of nearly a million clonal families! 3/7

December 17, 2024 at 4:01 PM
Our bodies make antibodies by evolving specific portions of their sequences to bind their target strongly and stably, resulting in a set of related sequences known as a clonal family. We leverage modern software and data to build a dataset of nearly a million clonal families! 3/7
SoTA methods search the space of sequences by iteratively suggesting mutations. But the space of antibodies is huge! CloneBO builds a prior on mutations that make strong and stable binders in our body to optimize antibodies in silico. 2/7

December 17, 2024 at 4:01 PM
SoTA methods search the space of sequences by iteratively suggesting mutations. But the space of antibodies is huge! CloneBO builds a prior on mutations that make strong and stable binders in our body to optimize antibodies in silico. 2/7
How do you go from a hit in your antibody screen to a suitable drug? Now introducing CloneBO: we optimize antibodies in the lab by teaching a generative model how we optimize them in our bodies!
w/ Nat Gruver, Yilun Kuang, Lily Li, @andrewgwils.bsky.social and the team at Big Hat! 1/7
w/ Nat Gruver, Yilun Kuang, Lily Li, @andrewgwils.bsky.social and the team at Big Hat! 1/7
December 17, 2024 at 4:01 PM
How do you go from a hit in your antibody screen to a suitable drug? Now introducing CloneBO: we optimize antibodies in the lab by teaching a generative model how we optimize them in our bodies!
w/ Nat Gruver, Yilun Kuang, Lily Li, @andrewgwils.bsky.social and the team at Big Hat! 1/7
w/ Nat Gruver, Yilun Kuang, Lily Li, @andrewgwils.bsky.social and the team at Big Hat! 1/7
New model trained on new dataset of nearly a million evolving antibody families at AIDrugX workshop Sunday at 4:20 pm (#76) #Neurips! Collab between @andrewgwils.bsky.social and BigHatBio. Stay tuned for full thread on how we used the model to optimize antibodies in the lab in coming days!

December 14, 2024 at 4:32 PM
New model trained on new dataset of nearly a million evolving antibody families at AIDrugX workshop Sunday at 4:20 pm (#76) #Neurips! Collab between @andrewgwils.bsky.social and BigHatBio. Stay tuned for full thread on how we used the model to optimize antibodies in the lab in coming days!