




Nilay Hazari
@nhazari.bsky.social
Chair and Professor of Chemistry at Yale University; PI of the Hazari group; Focus on organometallic, inorganic, and organic chemistry and catalysis.
How do transition metal hydrides transfer hydrides to organic substrates? Sometimes it can be predicted using Marcus theory. See pubs.acs.org/doi/full/10.... for our latest results on this topic. Thank you to Alex Miller and Zahid Ertem for all their help!

Elucidation of Marcus Relationships for Hydride Transfer Reactions Involving Transition Metal Hydrides
The rate of hydride transfer from three Ir hydride complexes of the type Cp*Ir(Rbpy)H+ (Cp* = C5Me5; Rbpy = 4,4′-R-2,2′-bipyridine, R = OMe, H, CO2Me) to six N-methylacridinium (RAcr+) acceptors with electronically different substituents in the 2- or 2,7-positions were measured. Using the thermodynamic hydricity of the donors and the hydride affinity of the acceptors the thermodynamic driving forces for hydride transfer were determined. Brønsted plots, which correlate kinetic and thermodynamic hydricity, demonstrate distinct linear free energy relationships for each complex, with different Brønsted α values. Thus, at the same driving force hydride transfer from Cp*Ir(OMebpy)H+ is faster than for Cp*Ir(bpy)H+ or Cp*Ir(CO2Mebpy)H+. Experimental and computational analyses are consistent with a concerted hydride transfer mechanism for all Ir complexes. As the thermodynamic driving force increases an earlier transition state is observed and all transition states also include π-stacking interactions between the donor and acceptor, which likely contribute to the different α values. The experimental data fits well to the Marcus model, enabling the determination of reorganization energies (λ) that range from 58 to 69 kcal mol–1. These are lower than λ values for hydride transfer reactions involving organic donors and acceptors. This work provides a rare example of the correlation of kinetic and thermodynamic hydricity using only experimental data and shows that hydride transfer reactions involving metal hydrides can follow Marcus theory. The findings offer insight into controlling metal-catalyzed hydride transfer reactions, which is valuable for designing improved systems for a range of transformations.
pubs.acs.org
August 26, 2025 at 12:40 AM
How do transition metal hydrides transfer hydrides to organic substrates? Sometimes it can be predicted using Marcus theory. See pubs.acs.org/doi/full/10.... for our latest results on this topic. Thank you to Alex Miller and Zahid Ertem for all their help!
New paper from the CHASE solar hub led by my friends Alex Miller (UNC) and Zahid Ertem (Brookhaven) on catalytic hydrogenation of a metal formyl to a metal carbonyl. Happy to play a small role in addressing this long standing challenge in organometallic chemistry. See pubs.acs.org/doi/full/10....
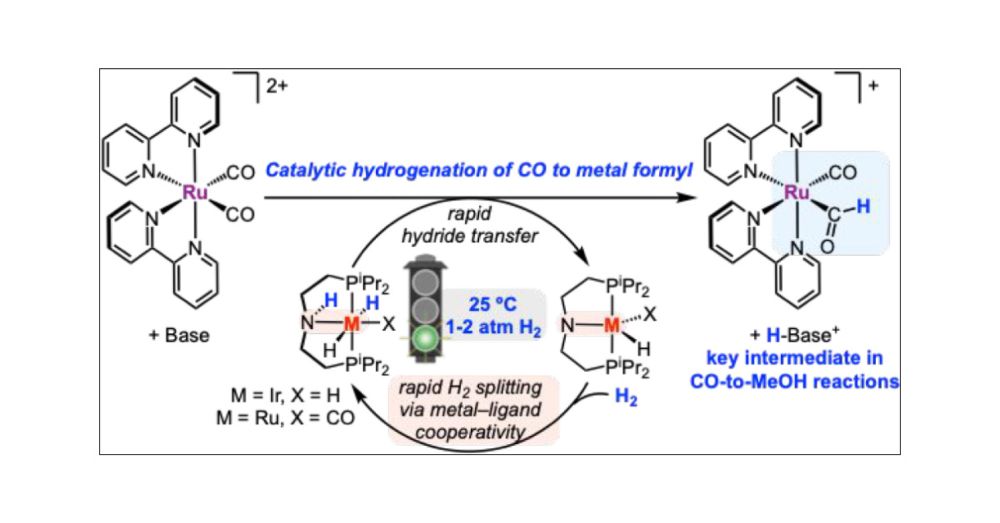
Catalytic Hydrogenation of a Ruthenium Carbonyl to Formyl Enabled by Metal–Ligand Cooperation
Metal formyl complexes are critical intermediates in the reduction of CO to valuable products such as methanol and higher alcohols/hydrocarbons, yet examples of formyl generation via the catalytic hydrogenation of transition metal carbonyl complexes under mild conditions are lacking. The catalytic hydrogenation of a ruthenium carbonyl complex with H2 to produce a formyl complex is reported here. Two classes of hydrogenation catalysts were compared: bis(diphosphine)-ligated complexes that proceed via termolecular H2 splitting with an external base and pincer-ligated complexes that proceed via an H2 splitting mechanism involving metal–ligand cooperativity. The hydride transfer and H2 splitting steps were evaluated for both classes of catalysts, revealing advantages for catalysts that utilize metal–ligand cooperativity and elucidating conditions to promote formyl generation. Only the pincer-ligated Ir and Ru complexes capable of reacting via pathways involving metal–ligand cooperativity were suitable for catalysis. Using 1–10 mol % of the catalysts (PNP)Ir(H)2 and (HPNP)Ru(H)2(CO) (PNP = (iPr2PC2H4)2N–), which use metal–ligand cooperation to activate H2, up to 10 turnovers or up to 71% yield were achieved for the conversion of [Ru(bpy)2(CO)2]2+ (bpy = 2,2′-bipyridine) to the formyl complex [Ru(bpy)2(CO)(CHO)]+. The Lewis acid B(C6F5)3 was required as an additive to achieve high yields of the formyl complex using (HPNP)Ru(H)2(CO) as a catalyst. The catalytic route avoids the use of expensive stoichiometric reagents, such as borohydride, instead generating metal formyls that are key intermediates in CO reduction schemes with H2 gas.
pubs.acs.org
July 22, 2025 at 1:07 PM
New paper from the CHASE solar hub led by my friends Alex Miller (UNC) and Zahid Ertem (Brookhaven) on catalytic hydrogenation of a metal formyl to a metal carbonyl. Happy to play a small role in addressing this long standing challenge in organometallic chemistry. See pubs.acs.org/doi/full/10....
A new General Chemistry video posted to the playlist: www.youtube.com/playlist?lis.... This one is about Modern Atomic Structure. Thank you to Yale's Poorvu Center for support.

General Chemistry Concepts Explained - YouTube
Professor Nilay Hazari of Yale University describes concepts from a standard first semester general chemistry taught at an American university. Each video is...
www.youtube.com
July 3, 2025 at 11:30 AM
A new General Chemistry video posted to the playlist: www.youtube.com/playlist?lis.... This one is about Modern Atomic Structure. Thank you to Yale's Poorvu Center for support.
A collaboration with Merck resulted in a paper on Ni/Ti dual catalyzed cross-electrophile coupling of aryl halides with unactivated alkyl chlorides. The key is to match the rate of alkyl radical generation by Ti with aryl halide activation by Ni. @acs.org. See: pubs.acs.org/doi/full/10....
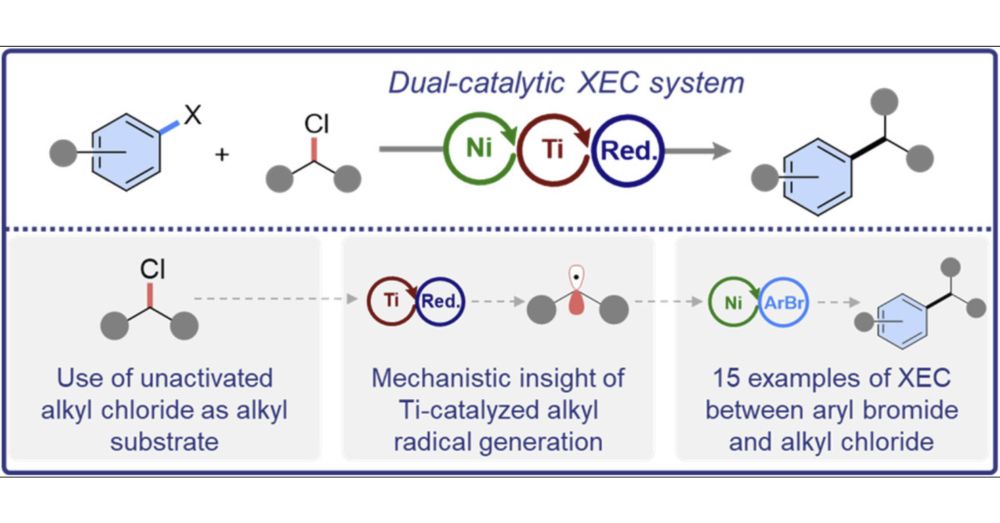
Ni/Ti Dual Catalyzed Cross-Electrophile Coupling between Unactivated Alkyl Chlorides and Aryl Halides
A dual Ni/Ti-catalyzed method for cross-electrophile coupling (XEC) of primary and secondary alkyl chlorides and aryl halides is described. This is a rare example of a thermal XEC reaction that directly couples unactivated alkyl chlorides, which are valuable substrates because of their accessibility and stability. Mechanistic studies indicate that the Ti catalyst, Cp*2TiIVCl2 (Cp* = pentamethyl-cyclopentadienyl), is crucial for activation of the alkyl chloride. Specifically, Cp*2TiIVCl2 undergoes reduction to form Cp*2TiIIICl, which was isolated and crystallographically characterized. Control experiments demonstrate that Cp*2TiIIICl reacts with primary, secondary, and tertiary alkyl chlorides to form alkyl radicals. While the Ni catalyst is not reactive enough to form alkyl radicals from alkyl chlorides directly, it is crucial for activating the aryl halide, resulting in the formation of an intermediate of the form (tBubpy)Ni(Ar)X (tBubpy = 4,4′-tBu2-2,2′-bipyridine; X = halide). Stoichiometric experiments showed that the (tBubpy)Ni(Ar)X intermediate captures alkyl radicals generated by the Ti catalyst and subsequently forms the organic XEC product. A key feature in the Ni/Ti dual catalyzed reaction is matching the rates of the Ni and Ti catalytic cycles, so that the rates of radical production and trapping are complementary. This can be achieved by varying the relative loadings of the Ni and Ti catalysts. It is expected that the strategy of using a second reactive catalyst to activate previously inert substrates in Ni-catalyzed XEC will be applicable to other challenging substrate classes.
pubs.acs.org
June 23, 2025 at 1:24 PM
A collaboration with Merck resulted in a paper on Ni/Ti dual catalyzed cross-electrophile coupling of aryl halides with unactivated alkyl chlorides. The key is to match the rate of alkyl radical generation by Ti with aryl halide activation by Ni. @acs.org. See: pubs.acs.org/doi/full/10....
New Flash Communication with my friend Wes Bernskoetter on Ir complexes with a modified PNP pincer ligand for carbon dioxide hydrogenation. See pubs.acs.org/doi/10.1021/.... @acs.org.
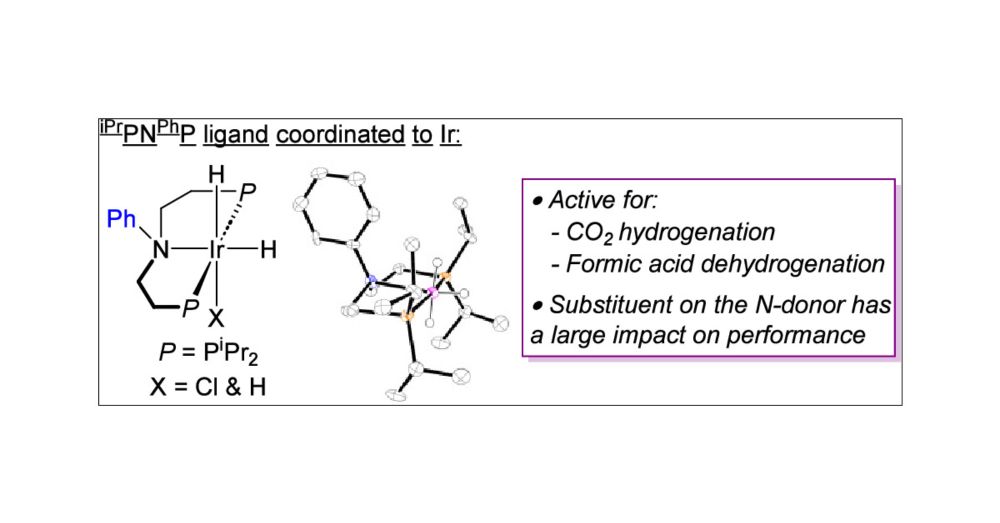
Flash Communication: Ir Complexes with a PhN(CH2CH2PiPr2)2 Pincer Ligand for Reversible CO2 Hydrogenation
The pincer ligand PhN(CH2CH2PiPr2)2 (iPrPNPhP) was treated with [Ir(coe)2(μ-Cl)]2 (coe = cyclooctene) under H2 to generate (iPrPNPhP)IrH2Cl (1). Reaction of 1 with LiBHEt3 formed (iPrPNPhP)IrH3 (2). T...
pubs.acs.org
June 21, 2025 at 3:01 PM
New Flash Communication with my friend Wes Bernskoetter on Ir complexes with a modified PNP pincer ligand for carbon dioxide hydrogenation. See pubs.acs.org/doi/10.1021/.... @acs.org.