




Aakash Naik
@naikaakash.bsky.social
Ph.D Student at @BAMResearch @MolecularXtal research group
Affiliated to @UniJena
Affiliated to @UniJena
Reposted by Aakash Naik

Interested in predicting magnetism in transition metal compounds? We have written a paper on how to use exchange heuristics in such models. We also show limits of current theoretical approaches.
Please find our preprint here.
doi.org/10.26434/che...
#compchemsky
Please find our preprint here.
doi.org/10.26434/che...
#compchemsky

Can simple exchange heuristics guide us in predicting magnetic properties of solids?
A popular heuristic derived from the Kanamori-Goodenough-Anderson rules of superexchange connects bond angles and magnetism in certain transition metal compounds. We evaluate the fulfillment of this h...
doi.org
August 15, 2025 at 12:29 PM
Interested in predicting magnetism in transition metal compounds? We have written a paper on how to use exchange heuristics in such models. We also show limits of current theoretical approaches.
Please find our preprint here.
doi.org/10.26434/che...
#compchemsky
Please find our preprint here.
doi.org/10.26434/che...
#compchemsky
Looking forward to presenting my PhD research at #S25MRS tomorrow! Excited to share my findings and engage in constructive discussions with the community
April 9, 2025 at 2:01 AM
Looking forward to presenting my PhD research at #S25MRS tomorrow! Excited to share my findings and engage in constructive discussions with the community
Reposted by Aakash Naik
CECAM school on automated ab initio calculations came to an end.
Nearly all teaching material including videos of our atomate2 school is already or will be online:
www.cecam.org/workshop-det...
#compchem
@virtualatoms.bsky.social @naikaakash.bsky.social and many more not on here 😀
Nearly all teaching material including videos of our atomate2 school is already or will be online:
www.cecam.org/workshop-det...
#compchem
@virtualatoms.bsky.social @naikaakash.bsky.social and many more not on here 😀
www.cecam.org
March 21, 2025 at 8:40 AM
CECAM school on automated ab initio calculations came to an end.
Nearly all teaching material including videos of our atomate2 school is already or will be online:
www.cecam.org/workshop-det...
#compchem
@virtualatoms.bsky.social @naikaakash.bsky.social and many more not on here 😀
Nearly all teaching material including videos of our atomate2 school is already or will be online:
www.cecam.org/workshop-det...
#compchem
@virtualatoms.bsky.social @naikaakash.bsky.social and many more not on here 😀
Reposted by Aakash Naik
🤖 Interested in automated DFT or ab initio calculations for crystals or molecules?
atomate2 could be your package!
doi.org/10.26434/che...
#compchem
atomate2 could be your package!
doi.org/10.26434/che...
#compchem

Atomate2: Modular workflows for materials science
High-throughput density functional theory (DFT) calculations have become a vital element of computational materials science, enabling materials screening, property database generation, and training of...
doi.org
January 22, 2025 at 7:28 PM
🤖 Interested in automated DFT or ab initio calculations for crystals or molecules?
atomate2 could be your package!
doi.org/10.26434/che...
#compchem
atomate2 could be your package!
doi.org/10.26434/che...
#compchem
Reposted by Aakash Naik

Meet autoplex – our approach to automated ML potential fitting, built jointly with @molecularxtal.bsky.social & team in Berlin! In this preprint, we focus on exploring structures and training potential models "from scratch" with the help of automated workflows: arxiv.org/abs/2412.16736

An automated framework for exploring and learning potential-energy surfaces
Machine learning has become ubiquitous in materials modelling and now routinely enables large-scale atomistic simulations with quantum-mechanical accuracy. However, developing machine-learned interato...
arxiv.org
January 7, 2025 at 6:44 PM
Meet autoplex – our approach to automated ML potential fitting, built jointly with @molecularxtal.bsky.social & team in Berlin! In this preprint, we focus on exploring structures and training potential models "from scratch" with the help of automated workflows: arxiv.org/abs/2412.16736
Reposted by Aakash Naik
New paper on "Chemical ordering and magnetism in face-centered cubic CrCoNi" alloy together with Sheuly Ghosh, Jörg Neugebauer and Fritz Körmann.
Katharina Ueltzen from our group used COHP-based bonding analysis to explain the magnetic ordering in the alloy 🥳
www.nature.com/articles/s41...
Katharina Ueltzen from our group used COHP-based bonding analysis to explain the magnetic ordering in the alloy 🥳
www.nature.com/articles/s41...
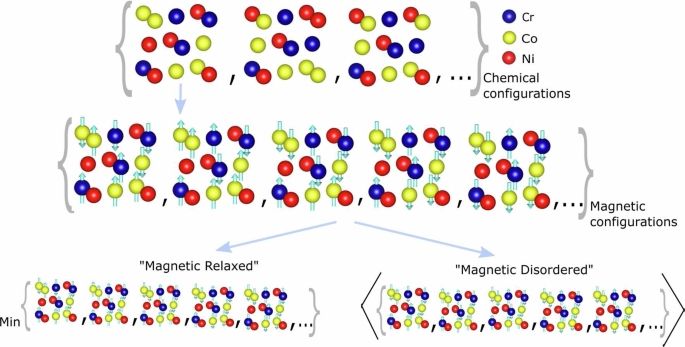
Chemical ordering and magnetism in face-centered cubic CrCoNi alloy - npj Computational Materials
npj Computational Materials - Chemical ordering and magnetism in face-centered cubic CrCoNi alloy
www.nature.com
December 20, 2024 at 11:40 AM
New paper on "Chemical ordering and magnetism in face-centered cubic CrCoNi" alloy together with Sheuly Ghosh, Jörg Neugebauer and Fritz Körmann.
Katharina Ueltzen from our group used COHP-based bonding analysis to explain the magnetic ordering in the alloy 🥳
www.nature.com/articles/s41...
Katharina Ueltzen from our group used COHP-based bonding analysis to explain the magnetic ordering in the alloy 🥳
www.nature.com/articles/s41...