




Marcel M
@mrclmllr.bsky.social
Postdoctoral Fellow at @thematterlab.bsky.social with @aspuru.bsky.social | PhD in Theoretical Chemistry | ex FCI scholar & Digital Chemistry @merckgroup.bsky.social
Reposted by Marcel M

Our second agent, El Agente Estructural (“Structural” in Spanish) is a multimodal, natural-language–driven agent for molecular geometry generation and manipulation.
🔗 www.arxiv.org/abs/2602.048...
[1/7]
🔗 www.arxiv.org/abs/2602.048...
[1/7]

February 6, 2026 at 7:45 PM
Our second agent, El Agente Estructural (“Structural” in Spanish) is a multimodal, natural-language–driven agent for molecular geometry generation and manipulation.
🔗 www.arxiv.org/abs/2602.048...
[1/7]
🔗 www.arxiv.org/abs/2602.048...
[1/7]
Reposted by Marcel M

🚀 JCIM: "Chemical Space Exploration with Artificial Mindless Molecules"
We present MindlessGen, an open-source tool for generating chemically diverse "mindless" molecules, and the MB2061 benchmark set with high-level reference data to test methods on unconventional systems.
doi.org/10.1021/acs....
We present MindlessGen, an open-source tool for generating chemically diverse "mindless" molecules, and the MB2061 benchmark set with high-level reference data to test methods on unconventional systems.
doi.org/10.1021/acs....

Chemical Space Exploration with Artificial “Mindless” Molecules
We introduce MindlessGen, a Python-based generator for creating chemically diverse, “mindless” molecules through random atomic placement and subsequent geometry optimization. Using this framework, we ...
doi.org
September 3, 2025 at 11:01 AM
🚀 JCIM: "Chemical Space Exploration with Artificial Mindless Molecules"
We present MindlessGen, an open-source tool for generating chemically diverse "mindless" molecules, and the MB2061 benchmark set with high-level reference data to test methods on unconventional systems.
doi.org/10.1021/acs....
We present MindlessGen, an open-source tool for generating chemically diverse "mindless" molecules, and the MB2061 benchmark set with high-level reference data to test methods on unconventional systems.
doi.org/10.1021/acs....
Reposted by Marcel M
We are delighted to announce that our perspective article, “Steering towards safe self-driving laboratories (SDLs),” has been accepted for publication in Nature Reviews Chemistry.
Link: www.nature.com/articles/s41...
Link: www.nature.com/articles/s41...

August 18, 2025 at 5:46 PM
We are delighted to announce that our perspective article, “Steering towards safe self-driving laboratories (SDLs),” has been accepted for publication in Nature Reviews Chemistry.
Link: www.nature.com/articles/s41...
Link: www.nature.com/articles/s41...
Reposted by Marcel M
Headed to Accelerate 2025? You cannot miss the presentations from The Matter Lab. We've got your back--here's your ultimate cheat sheet so you don't miss a thing.
#Accelerate2025 #AIinScience
[1/2]
#Accelerate2025 #AIinScience
[1/2]


August 8, 2025 at 6:28 PM
Headed to Accelerate 2025? You cannot miss the presentations from The Matter Lab. We've got your back--here's your ultimate cheat sheet so you don't miss a thing.
#Accelerate2025 #AIinScience
[1/2]
#Accelerate2025 #AIinScience
[1/2]
Reposted by Marcel M

You can now use g-xTB @grimmelab.bsky.social with ORCA via the ExtOpt feature! Check out our new tutorial and learn how to use it in GOAT, NEB-TS and more.
www.faccts.de/docs/orca/6....
#ORCAqc #FACCTs #gxTB #CompChem #QuantumChem
www.faccts.de/docs/orca/6....
#ORCAqc #FACCTs #gxTB #CompChem #QuantumChem
ORCA as External Optimizer - ORCA 6.1 TUTORIALS
www.faccts.de
June 26, 2025 at 8:32 AM
You can now use g-xTB @grimmelab.bsky.social with ORCA via the ExtOpt feature! Check out our new tutorial and learn how to use it in GOAT, NEB-TS and more.
www.faccts.de/docs/orca/6....
#ORCAqc #FACCTs #gxTB #CompChem #QuantumChem
www.faccts.de/docs/orca/6....
#ORCAqc #FACCTs #gxTB #CompChem #QuantumChem
📢 Update on our g-xTB release published on ChemRxiv:
We’ve uploaded a Linux executable of the current development version of g-xTB on GitHub, along with a simple usage guide:
🔗 github.com/grimme-lab/g...
⚠️ Please note:
This is a preliminary release — currently Linux-only, using numerical gradients.
We’ve uploaded a Linux executable of the current development version of g-xTB on GitHub, along with a simple usage guide:
🔗 github.com/grimme-lab/g...
⚠️ Please note:
This is a preliminary release — currently Linux-only, using numerical gradients.
June 24, 2025 at 1:07 PM
📢 Update on our g-xTB release published on ChemRxiv:
We’ve uploaded a Linux executable of the current development version of g-xTB on GitHub, along with a simple usage guide:
🔗 github.com/grimme-lab/g...
⚠️ Please note:
This is a preliminary release — currently Linux-only, using numerical gradients.
We’ve uploaded a Linux executable of the current development version of g-xTB on GitHub, along with a simple usage guide:
🔗 github.com/grimme-lab/g...
⚠️ Please note:
This is a preliminary release — currently Linux-only, using numerical gradients.
After years of development and preparatory works which you might have seen on this profile, a major milestone is achieved:
g-xTB marks not just an evolution, but a revolution in the capabilities of semiempirical quantum chemistry. Convince yourself! A thread.
🔗 chemrxiv.org/engage/chemr...
#compchem
g-xTB marks not just an evolution, but a revolution in the capabilities of semiempirical quantum chemistry. Convince yourself! A thread.
🔗 chemrxiv.org/engage/chemr...
#compchem

g-xTB: A General-Purpose Extended Tight-Binding Electronic Structure Method For the Elements H to Lr (Z=1–103)
We present g-xTB, a next-generation semi-empirical electronic structure method derived from tight-binding (TB) approximations to Kohn–Sham density functional theory (KS-DFT). Designed to bridge the ga...
chemrxiv.org
June 24, 2025 at 7:31 AM
After years of development and preparatory works which you might have seen on this profile, a major milestone is achieved:
g-xTB marks not just an evolution, but a revolution in the capabilities of semiempirical quantum chemistry. Convince yourself! A thread.
🔗 chemrxiv.org/engage/chemr...
#compchem
g-xTB marks not just an evolution, but a revolution in the capabilities of semiempirical quantum chemistry. Convince yourself! A thread.
🔗 chemrxiv.org/engage/chemr...
#compchem
Reposted by Marcel M

#RobSelects preprint of the week #ChemRxiv: Benchmarking density functional approximations with a systematic set of randomly generated molecules. #compchem https://doi.org/10.26434/chemrxiv-2025-rdsd0

Chemical Space Exploration with Artificial ”Mindless” Molecules
We introduce MindlessGen, a Python-based generator for creating chemically diverse, “mindless” molecules through random atomic placement and subsequent geometry optimization. Using this framework, we constructed the MB2061 benchmark set, containing 2061 molecules with high-level PNO-LCCSD(T)-F12 reference data for dissociation reactions. This set provides a challenging benchmark for testing, validation, and training of density functional approximations (DFAs), semiempirical methods, force fields, and machine learning potentials using molecular structures beyond the conventional chemical space. For DFAs, we initially hypothesized that highly parameterized functionals might perform poorly on this set. However, no consistent relationship between fitting strategy and accuracy was observed. A clear Jacob’s ladder trend emerges, with ωB97X-2 achieving the lowest mean absolute error (MAE) of 8.4 kcal·mol−1 and r²SCAN-3c offering a robust cost-efficient alternative (19.6 kcal·mol−1). Furthermore, we discuss the performance of selected semiempirical methods and contemporary machine learned interatomic potentials.
chemrxiv.org
June 18, 2025 at 8:38 AM
#RobSelects preprint of the week #ChemRxiv: Benchmarking density functional approximations with a systematic set of randomly generated molecules. #compchem https://doi.org/10.26434/chemrxiv-2025-rdsd0
Reposted by Marcel M

Check out our new EEQBC model!
It delivers accurate and robust atomic charges for all elements up to Z=103. By incorporating bond capacitors, we eliminate most artificial CT while preserving the simplicity and efficiency of classical charge equilibration:
doi.org/10.26434/che...
#compchem
It delivers accurate and robust atomic charges for all elements up to Z=103. By incorporating bond capacitors, we eliminate most artificial CT while preserving the simplicity and efficiency of classical charge equilibration:
doi.org/10.26434/che...
#compchem

The Bond Capacity Electronegativity Equilibration Charge Model (EEQBC) for the Elements Z=1–103
The accurate and efficient assignment of atomic partial charges is crucial for many applications in theoretical and computational chemistry, including polarizable force fields, dispersion corrections, a...
doi.org
March 7, 2025 at 12:35 PM
Check out our new EEQBC model!
It delivers accurate and robust atomic charges for all elements up to Z=103. By incorporating bond capacitors, we eliminate most artificial CT while preserving the simplicity and efficiency of classical charge equilibration:
doi.org/10.26434/che...
#compchem
It delivers accurate and robust atomic charges for all elements up to Z=103. By incorporating bond capacitors, we eliminate most artificial CT while preserving the simplicity and efficiency of classical charge equilibration:
doi.org/10.26434/che...
#compchem
Say Hello to the Bannwarth group at Bluesky and give them a follow for great science! 👋 @bannwarthlab.bsky.social 🚀🔬
February 14, 2025 at 6:25 PM
Say Hello to the Bannwarth group at Bluesky and give them a follow for great science! 👋 @bannwarthlab.bsky.social 🚀🔬
Reposted by Marcel M
Thank you for your question! While an energy expression in the context of density-corrected DFT can still be conceptually very inspiring, we are currently working on a “real” xTB successor, called g-xTB.
This plot about the accuracy of the barrier heights compared to DFT gives a good impression. 💡
This plot about the accuracy of the barrier heights compared to DFT gives a good impression. 💡

January 30, 2025 at 9:53 AM
Thank you for your question! While an energy expression in the context of density-corrected DFT can still be conceptually very inspiring, we are currently working on a “real” xTB successor, called g-xTB.
This plot about the accuracy of the barrier heights compared to DFT gives a good impression. 💡
This plot about the accuracy of the barrier heights compared to DFT gives a good impression. 💡
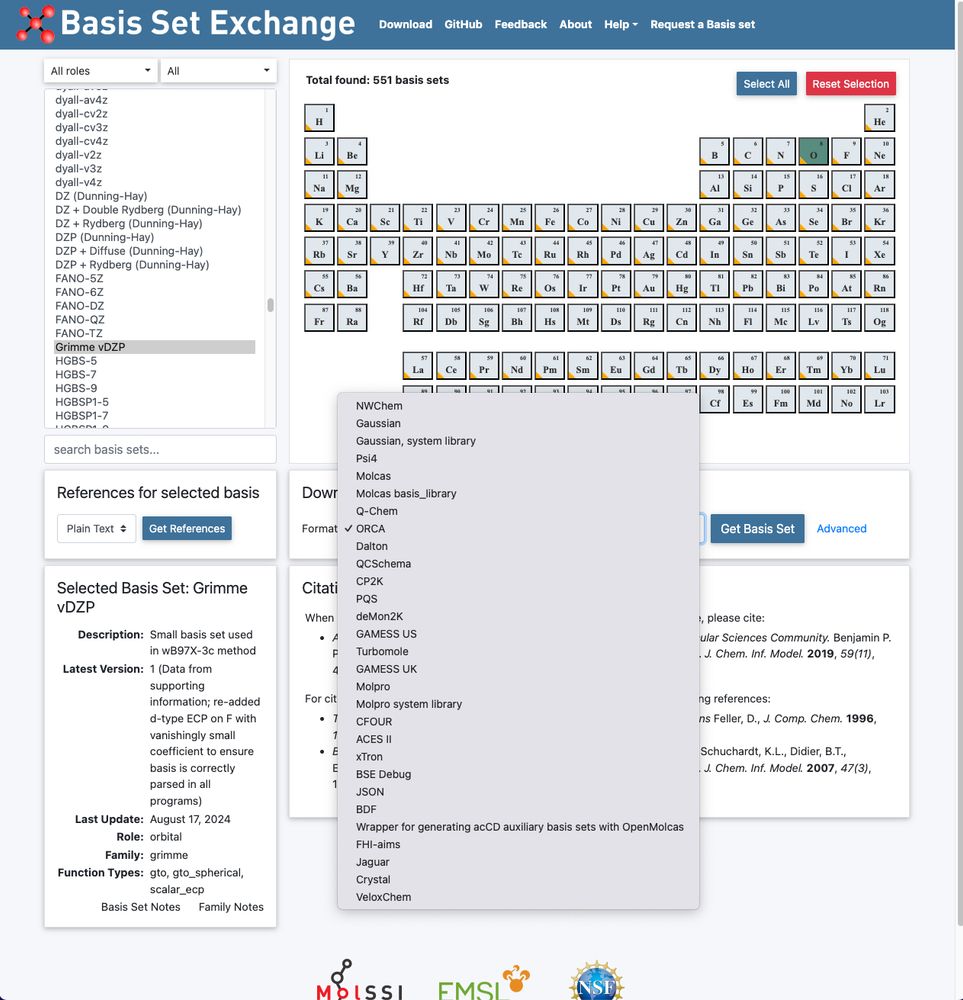
Our vDZP basis set utilized in the ⍵B97X-3c composite DFT method is now also available via www.basissetexchange.org (API-based: github.com/MolSSI-BSE/b...). 🎉
Many thanks to @Susi Lehtola & coworkers for jointly providing it there!
Many thanks to @Susi Lehtola & coworkers for jointly providing it there!
January 29, 2025 at 4:03 PM
Our vDZP basis set utilized in the ⍵B97X-3c composite DFT method is now also available via www.basissetexchange.org (API-based: github.com/MolSSI-BSE/b...). 🎉
Many thanks to @Susi Lehtola & coworkers for jointly providing it there!
Many thanks to @Susi Lehtola & coworkers for jointly providing it there!
Reposted by Marcel M

Happy to announce the release of @avogadro.cc 1.100 (not quite 2.0 yet) with a pile of new features, bug fixes, etc.
Can't fit the whole release notes here, but more on the forum:
discuss.avogadro.cc/t/avogadro-1...
Can't fit the whole release notes here, but more on the forum:
discuss.avogadro.cc/t/avogadro-1...
January 22, 2025 at 2:13 PM
Happy to announce the release of @avogadro.cc 1.100 (not quite 2.0 yet) with a pile of new features, bug fixes, etc.
Can't fit the whole release notes here, but more on the forum:
discuss.avogadro.cc/t/avogadro-1...
Can't fit the whole release notes here, but more on the forum:
discuss.avogadro.cc/t/avogadro-1...
I have created a timeline of semiempirical methods in quantum chemistry. Any thoughts, suggestions, or remarks on it? 💡 Have we missed anything?

January 22, 2025 at 12:22 PM
I have created a timeline of semiempirical methods in quantum chemistry. Any thoughts, suggestions, or remarks on it? 💡 Have we missed anything?
Reposted by Marcel M

Stefan Grimme receives the 2025 Chemistry Europe Award!
Learn more at https://buff.ly/3BXDVhO.
#ChemistryAward #ChemistryEurope
Learn more at https://buff.ly/3BXDVhO.
#ChemistryAward #ChemistryEurope

January 16, 2025 at 7:38 AM
Stefan Grimme receives the 2025 Chemistry Europe Award!
Learn more at https://buff.ly/3BXDVhO.
#ChemistryAward #ChemistryEurope
Learn more at https://buff.ly/3BXDVhO.
#ChemistryAward #ChemistryEurope
Important upgrade for the CEH charge model 📈
We’ve improved accuracy and robustness, and extended it for the actinides with f electrons in the valence space to cover elements all the way up to Z=103, together with the atom-in-molecule adaptive q-vSZP basis set! ⚛️ pubs.acs.org/doi/10.1021/...
We’ve improved accuracy and robustness, and extended it for the actinides with f electrons in the valence space to cover elements all the way up to Z=103, together with the atom-in-molecule adaptive q-vSZP basis set! ⚛️ pubs.acs.org/doi/10.1021/...
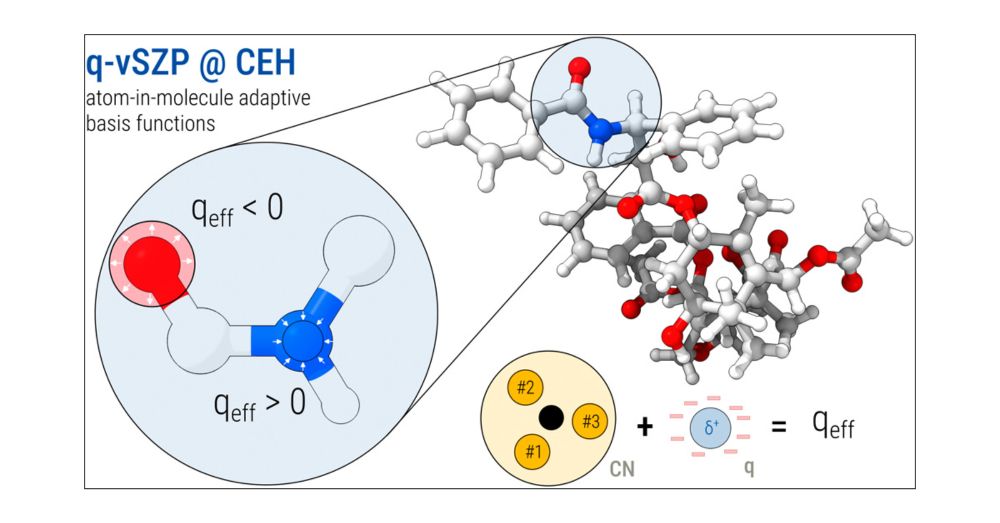
Advanced Charge Extended Hückel (CEH) Model and a Consistent Adaptive Minimal Basis Set for the Elements Z = 1–103
The Charge Extended Hückel (CEH) model, initially introduced for adaptive atomic orbital (AO) basis set construction (J. Chem. Phys. 2023, 159, 164108), has been significantly revised to enhance accur...
pubs.acs.org
December 17, 2024 at 10:17 AM
Important upgrade for the CEH charge model 📈
We’ve improved accuracy and robustness, and extended it for the actinides with f electrons in the valence space to cover elements all the way up to Z=103, together with the atom-in-molecule adaptive q-vSZP basis set! ⚛️ pubs.acs.org/doi/10.1021/...
We’ve improved accuracy and robustness, and extended it for the actinides with f electrons in the valence space to cover elements all the way up to Z=103, together with the atom-in-molecule adaptive q-vSZP basis set! ⚛️ pubs.acs.org/doi/10.1021/...
PTB (xtb-docs.readthedocs.io/en/latest/pt...) is finally available on all platforms in xtb>=6.7.0 (github.com/grimme-lab/xtb), including conda-forge – give it a try for electron densities at DFT-level with tight-binding speed and very accurate vibrational spectroscopy intensities! ⚡️ #compchem

December 12, 2024 at 1:54 PM
PTB (xtb-docs.readthedocs.io/en/latest/pt...) is finally available on all platforms in xtb>=6.7.0 (github.com/grimme-lab/xtb), including conda-forge – give it a try for electron densities at DFT-level with tight-binding speed and very accurate vibrational spectroscopy intensities! ⚡️ #compchem
Good news for all macOS users and everybody else using brew: I've updated all @grimmelab.bsky.social and -related software packages (e.g., 'xtb') to their latest release! 👨🏼💻🚀 github.com/grimme-lab/homebrew-qc
#brew #macos #xtb
#brew #macos #xtb
GitHub - grimme-lab/homebrew-qc: Brew formulas for xtb and related quantum chemistry programs
Brew formulas for xtb and related quantum chemistry programs - grimme-lab/homebrew-qc
github.com
November 21, 2024 at 3:28 PM
Good news for all macOS users and everybody else using brew: I've updated all @grimmelab.bsky.social and -related software packages (e.g., 'xtb') to their latest release! 👨🏼💻🚀 github.com/grimme-lab/homebrew-qc
#brew #macos #xtb
#brew #macos #xtb
Interested in a simple, lightweight method providing atomic charges and (atomic) dipole moments? According to our findings, CEH outperforms simple charge equilibration methods while still being way faster than typical SQM/tight-binding methods!
Details at: pubs.aip.org/aip/jcp/arti...
Details at: pubs.aip.org/aip/jcp/arti...

An atom-in-molecule adaptive polarized valence single-ζ atomic orbital basis for electronic structu...
Many low-cost or semiempirical quantum mechanical-based electronic structure methods suffer from the use of unpolarized minimal atomic orbital (AO) basis sets.
pubs.aip.org
October 31, 2023 at 8:32 AM
Interested in a simple, lightweight method providing atomic charges and (atomic) dipole moments? According to our findings, CEH outperforms simple charge equilibration methods while still being way faster than typical SQM/tight-binding methods!
Details at: pubs.aip.org/aip/jcp/arti...
Details at: pubs.aip.org/aip/jcp/arti...