



Konrad
@konradjk.bsky.social
Genomicist, computational biologist. Assistant professor @ MGH, HMS. Associate member @ Broad Institute
https://klab.is
https://klab.is
A project many years in the process, we’re pleased to present our work on multi-ancestry meta-analysis across a boatload of traits in the UK Biobank: www.nature.com/articles/s41...

Pan-UK Biobank genome-wide association analyses enhance discovery and resolution of ancestry-enriched effects - Nature Genetics
Genome-wide analyses for 7,266 traits leveraging data from several genetic ancestry groups in UK Biobank identify new associations and enhance resources for interpreting risk variants across diverse p...
www.nature.com
September 18, 2025 at 5:25 PM
A project many years in the process, we’re pleased to present our work on multi-ancestry meta-analysis across a boatload of traits in the UK Biobank: www.nature.com/articles/s41...
We’ve put up summary statistics for over 3,000 traits in the All of Us resource, and a shiny new browser alongside it! Explore your favorite gene or phenotype here: allbyall.researchallofus.org #ASHG24
All by All
The All by All browser maps known and novel associations between genotypes and phenotypes using data contributed by All of Us Research Program participants as of July 1, 2022. All by All encompasses a...
allbyall.researchallofus.org
November 8, 2024 at 8:32 PM
We’ve put up summary statistics for over 3,000 traits in the All of Us resource, and a shiny new browser alongside it! Explore your favorite gene or phenotype here: allbyall.researchallofus.org #ASHG24
We have a new preprint that we’d love feedback on! We benchmarked a bunch of variant scoring methods to figure out what they were actually doing, and how they performed across selection regimes: www.biorxiv.org/content/10.1...

Variant scoring performance across selection regimes depends on variant-to-gene and gene-to-disease components
bioRxiv - the preprint server for biology, operated by Cold Spring Harbor Laboratory, a research and educational institution
www.biorxiv.org
September 20, 2024 at 3:47 PM
We have a new preprint that we’d love feedback on! We benchmarked a bunch of variant scoring methods to figure out what they were actually doing, and how they performed across selection regimes: www.biorxiv.org/content/10.1...
Welcome new followers (and thanks @michelnivard.bsky.social)! I’m loving the critical mass, and to celebrate, I’ll post some exciting new content (my first time posting here and not on the the other site)
September 20, 2024 at 3:46 PM
Welcome new followers (and thanks @michelnivard.bsky.social)! I’m loving the critical mass, and to celebrate, I’ll post some exciting new content (my first time posting here and not on the the other site)
Reposted by Konrad

Recently out on #bioRxiv: our updated approach to identify regional variability in missense mutation intolerance (“constraint”) in protein-coding genes using the gnomAD database.
www.biorxiv.org/content/10.1...
1/10
www.biorxiv.org/content/10.1...
1/10
April 19, 2024 at 11:34 PM
Recently out on #bioRxiv: our updated approach to identify regional variability in missense mutation intolerance (“constraint”) in protein-coding genes using the gnomAD database.
www.biorxiv.org/content/10.1...
1/10
www.biorxiv.org/content/10.1...
1/10
As genomic analyses scale to millions of exomes/genomes, we need a scalable infrastructure to process/QC/handle these data while retaining all the metrics needed for downstream analysis. A new preprint from the Hail team proposes a way to do this! Comments welcome: www.biorxiv.org/content/10.1...

The Scalable Variant Call Representation: Enabling Genetic Analysis Beyond One Million Genomes
bioRxiv - the preprint server for biology, operated by Cold Spring Harbor Laboratory, a research and educational institution
www.biorxiv.org
January 31, 2024 at 3:40 PM
As genomic analyses scale to millions of exomes/genomes, we need a scalable infrastructure to process/QC/handle these data while retaining all the metrics needed for downstream analysis. A new preprint from the Hail team proposes a way to do this! Comments welcome: www.biorxiv.org/content/10.1...
Thrilled to have our work on gnomAD out in print at Nature today. With 76K genomes, we can look beyond the coding genome and into the non-coding genome to find regions important for human disease idp.nature.com/authorize?re...
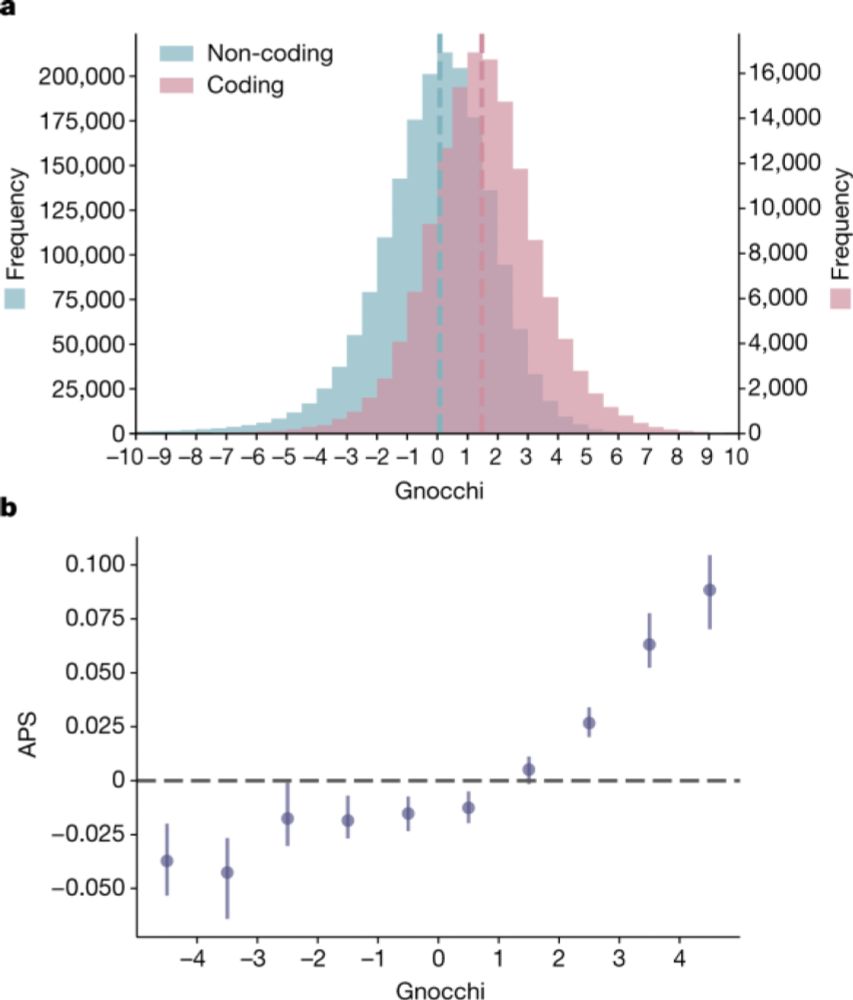
A genomic mutational constraint map using variation in 76,156 human genomes - Nature
A genomic constraint map for the human genome constructed using data from 76,156 human genomes from the Genome Aggregation Database shows that non-coding constrained regions are enriched for regulator...
idp.nature.com
December 6, 2023 at 5:10 PM
Thrilled to have our work on gnomAD out in print at Nature today. With 76K genomes, we can look beyond the coding genome and into the non-coding genome to find regions important for human disease idp.nature.com/authorize?re...
Excited for my first post on this new site to share our work in print at AJHG using variant call data to estimate DNA contamination. As our sample sizes get into the millions of genomes, we need methods like this to efficiently process and quality control the data authors.elsevier.com/c/1i8PAgeX6LB~
November 28, 2023 at 7:34 PM
Excited for my first post on this new site to share our work in print at AJHG using variant call data to estimate DNA contamination. As our sample sizes get into the millions of genomes, we need methods like this to efficiently process and quality control the data authors.elsevier.com/c/1i8PAgeX6LB~