



Jan Řezáč
@jrezac.bsky.social
Computational chemist at @iocbprague.bsky.social
The workshop "Quantum Chemistry for Drug Design: From Theory to Applications", which we organized at IOCB Prague, has just concluded. Leading academics and pharmaceutical industry practitioners came together to share their knowledge and insights.
Thanks to everybody who made it happen!
Thanks to everybody who made it happen!

September 12, 2025 at 10:14 AM
The workshop "Quantum Chemistry for Drug Design: From Theory to Applications", which we organized at IOCB Prague, has just concluded. Leading academics and pharmaceutical industry practitioners came together to share their knowledge and insights.
Thanks to everybody who made it happen!
Thanks to everybody who made it happen!
Reposted by Jan Řezáč

Day 3 opened with Kenneth Atz @Roche tackling the holy grail of #CADD: P-L binding #affinity prediction. By reframing limits of data & models, we can focus on the next solvable challenges - a sharp reminder of complexity & progress ahead.
#CECAM @cecamevents.bsky.social @iocbprague.bsky.social
#CECAM @cecamevents.bsky.social @iocbprague.bsky.social

September 10, 2025 at 9:31 AM
Day 3 opened with Kenneth Atz @Roche tackling the holy grail of #CADD: P-L binding #affinity prediction. By reframing limits of data & models, we can focus on the next solvable challenges - a sharp reminder of complexity & progress ahead.
#CECAM @cecamevents.bsky.social @iocbprague.bsky.social
#CECAM @cecamevents.bsky.social @iocbprague.bsky.social
Reposted by Jan Řezáč
We opened Day 2 of our #CECAM flagship workshop in Prague with the CECAM director Andrea Cavalli, highlighting steered #MD, dynamical docking & the complexity of binding energetics, and the challenges ahead. 🚀
#CECAMinPrague @iocbprague.bsky.social @cecamevents.bsky.social @iocbtech.bsky.social
#CECAMinPrague @iocbprague.bsky.social @cecamevents.bsky.social @iocbtech.bsky.social

September 9, 2025 at 11:21 AM
We opened Day 2 of our #CECAM flagship workshop in Prague with the CECAM director Andrea Cavalli, highlighting steered #MD, dynamical docking & the complexity of binding energetics, and the challenges ahead. 🚀
#CECAMinPrague @iocbprague.bsky.social @cecamevents.bsky.social @iocbtech.bsky.social
#CECAMinPrague @iocbprague.bsky.social @cecamevents.bsky.social @iocbtech.bsky.social
PM6-ML, our semiempirical quantum-mechanical #CompChem method with machine learning correction (see paper: pubs.acs.org/doi/10.1021/...), is now also available as an Atomic Simulation Environment (ASE) calculator.
github.com/Honza-R/PM6-...
github.com/Honza-R/PM6-...

July 15, 2025 at 11:16 AM
PM6-ML, our semiempirical quantum-mechanical #CompChem method with machine learning correction (see paper: pubs.acs.org/doi/10.1021/...), is now also available as an Atomic Simulation Environment (ASE) calculator.
github.com/Honza-R/PM6-...
github.com/Honza-R/PM6-...
I added the g-xTB #compchem method, just introduced by @grimmelab.bsky.social, to our protein-ligand interaction energy benchmarking. With an average error of less than 5% in the PLA 15 dataset, it is the most accurate semiempirical QM method to date (when ML is not considered).

July 7, 2025 at 12:51 PM
I added the g-xTB #compchem method, just introduced by @grimmelab.bsky.social, to our protein-ligand interaction energy benchmarking. With an average error of less than 5% in the PLA 15 dataset, it is the most accurate semiempirical QM method to date (when ML is not considered).
1/2 Lying is not the same as hallucinating. I asked an LLM to write a script to fetch data from a public API. After a couple of iterations, during which I fixed the issues and the AI apologized, it started telling me that the code was correct, but that it was having trouble connecting to the API.
July 2, 2025 at 9:39 AM
1/2 Lying is not the same as hallucinating. I asked an LLM to write a script to fetch data from a public API. After a couple of iterations, during which I fixed the issues and the AI apologized, it started telling me that the code was correct, but that it was having trouble connecting to the API.
Reposted by Jan Řezáč

🚀 Benchmark paper out!
How well do DFT, semiempirical & ML methods model proton transfer?
✅ DFT performs well, except with N-groups
❌ Pure ML struggles (though ORB v3 shows big gains)
🔥 PM6-ML Δ-learning excels, even in QM/MM setups!
Check it out: pubs.acs.org/doi/10.1021/...
How well do DFT, semiempirical & ML methods model proton transfer?
✅ DFT performs well, except with N-groups
❌ Pure ML struggles (though ORB v3 shows big gains)
🔥 PM6-ML Δ-learning excels, even in QM/MM setups!
Check it out: pubs.acs.org/doi/10.1021/...

Benchmark of Approximate Quantum Chemical and Machine Learning Potentials for Biochemical Proton Transfer Reactions
Proton transfer reactions are among the most common chemical transformations and are central to enzymatic catalysis and bioenergetic processes. Their mechanisms are often investigated using DFT or approximate quantum chemical methods, whose accuracy directly impacts the reliability of the simulations. Here, a comprehensive set of semiempirical molecular orbital and tight-binding DFT approaches, along with recently developed machine learning (ML) potentials, are benchmarked against high-level MP2 reference data for a curated set of proton transfer reactions representative of biochemical systems. Relative energies, geometries, and dipole moments are evaluated for isolated reactions. Microsolvated reactions are also simulated using a hybrid QM/MM partition. Traditional DFT methods offer high accuracy in general but show markedly larger deviations for proton transfers involving nitrogen-containing groups. Among approximate models, RM1, PM6, PM7, DFTB2-NH, DFTB3, and GFN2-xTB show reasonable accuracy across properties, though their performance varies by chemical group. The ML-corrected (Δ-learning) model PM6-ML improves accuracy for all properties and chemical groups and transfers well to QM/MM simulations. Conversely, standalone ML potentials perform poorly for most reactions. These results provide a basis for evaluating approximate methods and selecting potentials for proton transfer simulations in complex environments.
pubs.acs.org
July 1, 2025 at 12:37 PM
🚀 Benchmark paper out!
How well do DFT, semiempirical & ML methods model proton transfer?
✅ DFT performs well, except with N-groups
❌ Pure ML struggles (though ORB v3 shows big gains)
🔥 PM6-ML Δ-learning excels, even in QM/MM setups!
Check it out: pubs.acs.org/doi/10.1021/...
How well do DFT, semiempirical & ML methods model proton transfer?
✅ DFT performs well, except with N-groups
❌ Pure ML struggles (though ORB v3 shows big gains)
🔥 PM6-ML Δ-learning excels, even in QM/MM setups!
Check it out: pubs.acs.org/doi/10.1021/...
I'm at the WATOC #CompChem conference in Oslo. Machine learning is everywhere, but the hottest news so far is the new g-xTB method by @grimmelab.bsky.social . The results presented today are truly impressive. I'm already running first calculations on our biomolecular systems...
June 26, 2025 at 9:39 AM
I'm at the WATOC #CompChem conference in Oslo. Machine learning is everywhere, but the hottest news so far is the new g-xTB method by @grimmelab.bsky.social . The results presented today are truly impressive. I'm already running first calculations on our biomolecular systems...
Our main topic is applying #compchem to protein-ligand interactions in #CADD. We just published a related article about using our semiempirical #QM methodology to analyze protein-protein interactions of the insulin receptor.
pubs.acs.org/doi/full/10....
pubs.acs.org/doi/full/10....

Multiscale Computational Protocols for Accurate Residue Interactions at the Flexible Insulin–Receptor Interface
The quantitative characterization of residue contributions to protein–protein binding across extensive flexible interfaces poses a significant challenge for biophysical computations. It is attributabl...
pubs.acs.org
May 20, 2025 at 11:05 AM
Our main topic is applying #compchem to protein-ligand interactions in #CADD. We just published a related article about using our semiempirical #QM methodology to analyze protein-protein interactions of the insulin receptor.
pubs.acs.org/doi/full/10....
pubs.acs.org/doi/full/10....
PM6-ML, our latest method that aims for quantum-chemical accuracy in large biomolecular systems, has a third implementation. In addition to MOPAC-ML and Cuby4, PM6-ML is now available in pDynamo3, where it can be used for QM/MM calculations: github.com/pdynamo/pDyn....

GitHub - pdynamo/pDynamo3: The pDynamo molecular modeling and simulation program
The pDynamo molecular modeling and simulation program - pdynamo/pDynamo3
github.com
May 19, 2025 at 6:47 AM
PM6-ML, our latest method that aims for quantum-chemical accuracy in large biomolecular systems, has a third implementation. In addition to MOPAC-ML and Cuby4, PM6-ML is now available in pDynamo3, where it can be used for QM/MM calculations: github.com/pdynamo/pDyn....
A humble #compchem contribution to a great experimental #medchem work ranging from novel synthesis protocol to in vivo models. We applied our SQM-based scoring to interpret the interaction of the novel inhibitors with the protein.

#research #medchem #antifungals
On-Resin Assembly of Macrocyclic Inhibitors of Cryptococcus neoformans May1: A Pathway to Potent Antifungal Agents (Kryštůfek et al.) – @pubs.acs.org J. Med. Chem.: doi.org/10.1021/acs....
@iocbprague.bsky.social @czechacademy.bsky.social @imgprague.bsky.social
On-Resin Assembly of Macrocyclic Inhibitors of Cryptococcus neoformans May1: A Pathway to Potent Antifungal Agents (Kryštůfek et al.) – @pubs.acs.org J. Med. Chem.: doi.org/10.1021/acs....
@iocbprague.bsky.social @czechacademy.bsky.social @imgprague.bsky.social

May 12, 2025 at 11:15 AM
Our PM6-ML method, a semiempirical QM method with ML correction, works well for proton transfer reactions - despite not having been trained for that. The new implementation reported in the preprint allows its use in QM/MM biomolecular simulations.
Pre-print alert!🚨 #CompChem
How do DFT, semiempirical & ML potentials handle proton transfers? ML-only performs poorly, but Δ-learning in PM6-ML (by @jrezac.bsky.social) shines, even in a hybrid QM/MM partition! DFT works well except for N-groups. Check it out:
chemrxiv.org/engage/chemr...
How do DFT, semiempirical & ML potentials handle proton transfers? ML-only performs poorly, but Δ-learning in PM6-ML (by @jrezac.bsky.social) shines, even in a hybrid QM/MM partition! DFT works well except for N-groups. Check it out:
chemrxiv.org/engage/chemr...

May 5, 2025 at 12:08 PM
Our PM6-ML method, a semiempirical QM method with ML correction, works well for proton transfer reactions - despite not having been trained for that. The new implementation reported in the preprint allows its use in QM/MM biomolecular simulations.
Reposted by Jan Řezáč
🚀 Exciting news! We're organizing a @cecamevents.bsky.social Flagship Workshop on Quantum Chemistry in Drug Design in Prague, Sept 8–10, 2025!
Join top experts from academia & industry. Few spots left for contributed talks!
📢 Apply now: www.cecam.org/workshop-det...
#compchem #cadd #QM #CECAM
Join top experts from academia & industry. Few spots left for contributed talks!
📢 Apply now: www.cecam.org/workshop-det...
#compchem #cadd #QM #CECAM
CECAM - Quantum Chemistry for Drug Design: From Theory to Applications
www.cecam.org
March 24, 2025 at 12:16 PM
🚀 Exciting news! We're organizing a @cecamevents.bsky.social Flagship Workshop on Quantum Chemistry in Drug Design in Prague, Sept 8–10, 2025!
Join top experts from academia & industry. Few spots left for contributed talks!
📢 Apply now: www.cecam.org/workshop-det...
#compchem #cadd #QM #CECAM
Join top experts from academia & industry. Few spots left for contributed talks!
📢 Apply now: www.cecam.org/workshop-det...
#compchem #cadd #QM #CECAM
Our new preprint - discussing the advantages and disadvantages of single-structure protein-ligand scoring (including our SQM2.20) in comparison to a wide range of MD-based methods.
doi.org/10.26434/che...
doi.org/10.26434/che...

Comparative Analysis of Quantum-Mechanical and standard Single-Structure Protein-Ligand Scoring Functions with MD-Based Free Energy Calculations
Single-structure scoring functions have been considered inferior to expensive ensemble free energy methods in predicting protein-ligand affinities. We are revisiting this dogma with the recently devel...
doi.org
March 17, 2025 at 8:18 AM
Our new preprint - discussing the advantages and disadvantages of single-structure protein-ligand scoring (including our SQM2.20) in comparison to a wide range of MD-based methods.
doi.org/10.26434/che...
doi.org/10.26434/che...
A perspective on the importance (and the lack of) reliable benchmarks for structure-based computer-aided drug design methods - with a contribution of @adampecina.bsky.social from my group
New Perspective on Community Benchmarking in Structure-Based Drug Design (SBDD)!
#SBDD predictions need reliable benchmarks - diverse targets, high-quality affinity & structural data, and blinded validation. Let’s make it happen!
🔗 Read more: doi.org/10.1021/acs....
#DrugDiscovery #CompChem
#SBDD predictions need reliable benchmarks - diverse targets, high-quality affinity & structural data, and blinded validation. Let’s make it happen!
🔗 Read more: doi.org/10.1021/acs....
#DrugDiscovery #CompChem
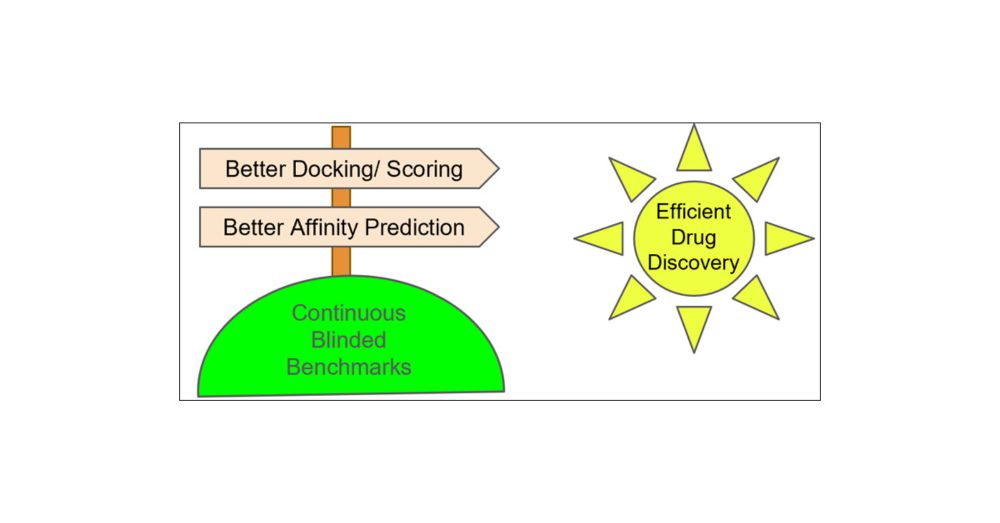
The Need for Continuing Blinded Pose- and Activity Prediction Benchmarks
Computational tools for structure-based drug design (SBDD) are widely used in drug discovery and can provide valuable insights to advance projects in an efficient and cost-effective manner. However, d...
doi.org
March 10, 2025 at 12:37 PM
A perspective on the importance (and the lack of) reliable benchmarks for structure-based computer-aided drug design methods - with a contribution of @adampecina.bsky.social from my group
We're organizing a CECAM workshop in September. If you're interested in QM calculations for drug design, apply and join us in Prague:
www.cecam.org/workshop-det...
www.cecam.org/workshop-det...
January 15, 2025 at 11:49 AM
We're organizing a CECAM workshop in September. If you're interested in QM calculations for drug design, apply and join us in Prague:
www.cecam.org/workshop-det...
www.cecam.org/workshop-det...
Our first paper of 2025: Δ-ML potential combining PM6 and a ML correction. Machine learning is doing wonders for correcting issues in PM6 that we could not fix any other way.
pubs.acs.org/doi/10.1021/...
pubs.acs.org/doi/10.1021/...

PM6-ML: The Synergy of Semiempirical Quantum Chemistry and Machine Learning Transformed into a Practical Computational Method
Machine learning (ML) methods offer a promising route to the construction of universal molecular potentials with high accuracy and low computational cost. It is becoming evident that integrating physi...
pubs.acs.org
January 13, 2025 at 8:30 AM
Our first paper of 2025: Δ-ML potential combining PM6 and a ML correction. Machine learning is doing wonders for correcting issues in PM6 that we could not fix any other way.
pubs.acs.org/doi/10.1021/...
pubs.acs.org/doi/10.1021/...