




Reza Maroofian
@reza-maroofian.bsky.social
Geneticist at UCL Queen Square Institute of Neurology @UCLIoN. Interested in Rare Diseases, Medical Genetics, Neurogenetics & Genomic Medicine.
Our new study on NDUFA9-related disorder builds on our earlier work on NDUFA11/12/13, revealing overlapping yet distinct clinical features that only emerge from larger, well-phenotyped cohorts. Together, these findings help define the evolving landscape of mitochondrial complex I–related disease.
Magrinelli et al. define the pheno-genotypic spectrum of NDUFA9-related mitochondrial disease in 12 cases. Biallelic NDUFA9 variants cause complex I deficiency, with symptoms from neurodevelopmental delay with dystonia to isolated dystonia with basal ganglia MRI abnormalities 👉 buff.ly/t5XWVkv

November 20, 2025 at 8:02 AM
Our new study on NDUFA9-related disorder builds on our earlier work on NDUFA11/12/13, revealing overlapping yet distinct clinical features that only emerge from larger, well-phenotyped cohorts. Together, these findings help define the evolving landscape of mitochondrial complex I–related disease.
We delineate GOT2 deficiency, expanding the malate–aspartate shuttle (MAS) spectrum as a developmental & epileptic encephalopathy (DEE). It clarifies routes to diagnosis/therapy, placing GOT2 alongside rare recessive DEEs from SLC25A12 & MDH1/2 as inborn MAS disorders. @uclqsion.bsky.social

GOT2 deficiency causes progressive neurodevelopmental disorder with epilepsy, microcephaly & white matter abnormalities. Low aspartate & high G3P are biomarkers. Serine/pyridoxine helped control seizures in 86%. Pyruvate enables metabolic rescue. 🧠🧬 bit.ly/4nd90iQ

October 26, 2025 at 11:14 PM
We delineate GOT2 deficiency, expanding the malate–aspartate shuttle (MAS) spectrum as a developmental & epileptic encephalopathy (DEE). It clarifies routes to diagnosis/therapy, placing GOT2 alongside rare recessive DEEs from SLC25A12 & MDH1/2 as inborn MAS disorders. @uclqsion.bsky.social
Reposted by Reza Maroofian

📣New from Pujol & colleagues!
📄Bi-allelic variants in the ribosomal protein RPS6KC1 cause a complex neurodevelopmental disorder
📄Bi-allelic variants in the ribosomal protein RPS6KC1 cause a complex neurodevelopmental disorder

Bi-allelic variants in the ribosomal protein RPS6KC1 cause a complex neurodevelopmental disorder
Bi-allelic variants in RPS6KC1 cause a neurodevelopmental disorder with features overlapping
with Coffin-Lowry syndrome. Functional studies reveal impaired ribosomal protein synthesis,
disrupted lipid...
www.cell.com
October 22, 2025 at 5:19 PM
📣New from Pujol & colleagues!
📄Bi-allelic variants in the ribosomal protein RPS6KC1 cause a complex neurodevelopmental disorder
📄Bi-allelic variants in the ribosomal protein RPS6KC1 cause a complex neurodevelopmental disorder
Our EPG5 study shows a continuum: early neurodevelopmental disruption and adult-onset neurodegeneration, both tied by autophagy defects. Like GBA1, PLA2G6, WDR45 & SYNJ1, PSMF1, EPG5 links rare paediatric disorders to adult PD/dementia. Rare informing common. bit.ly/432N7LT @UCLIoN @UCLBrainScience

Mutations in the Key Autophagy Tethering Factor EPG5 Link Neurodevelopmental and Neurodegenerative Disorders Including Early‐Onset Parkinsonism
Objective Autophagy is a fundamental biological pathway with vital roles in intracellular homeostasis. During autophagy, defective cargoes including mitochondria are targeted to lysosomes for cleara...
bit.ly
October 9, 2025 at 10:04 AM
Our EPG5 study shows a continuum: early neurodevelopmental disruption and adult-onset neurodegeneration, both tied by autophagy defects. Like GBA1, PLA2G6, WDR45 & SYNJ1, PSMF1, EPG5 links rare paediatric disorders to adult PD/dementia. Rare informing common. bit.ly/432N7LT @UCLIoN @UCLBrainScience
Reposted by Reza Maroofian

New international collaborative work incl. our group @erasmusmc.bsky.social on MACF1 published @ajhgnews.bsky.social
A clinical and genotype-phenotype analysis of MACF1 variants www.sciencedirect.com/science/arti...
#raredisease #genetics
A clinical and genotype-phenotype analysis of MACF1 variants www.sciencedirect.com/science/arti...
#raredisease #genetics

A clinical and genotype-phenotype analysis of MACF1 variants
Microtubule-actin cross-linking factor 1 (MACF1) is a large protein of the spectraplakin family, which is essential for brain development. MACF1 inter…
www.sciencedirect.com
September 19, 2025 at 7:34 PM
New international collaborative work incl. our group @erasmusmc.bsky.social on MACF1 published @ajhgnews.bsky.social
A clinical and genotype-phenotype analysis of MACF1 variants www.sciencedirect.com/science/arti...
#raredisease #genetics
A clinical and genotype-phenotype analysis of MACF1 variants www.sciencedirect.com/science/arti...
#raredisease #genetics
Reposted by Reza Maroofian

Combinatorial transcriptional regulation establishes subtype-appropriate synaptic properties in auditory neurons

Combinatorial transcriptional regulation establishes subtype-appropriate synaptic properties in auditory neurons
Bastille and colleagues demonstrate that two closely related transcription factors, c-Maf and Mafb, have both redundant and independent effects on the diversification and functional differentiation of peripheral auditory neurons. Their data suggest that varying combinations of c-Maf and Mafb generate nuanced differences in synaptic structure and function.
dlvr.it
June 10, 2025 at 12:36 PM
Combinatorial transcriptional regulation establishes subtype-appropriate synaptic properties in auditory neurons
Our new study characterises ELFN1 deficiency as a novel autosomal recessive neurodevelopmental disorder marked by epilepsy, GDD/ID, & movement disorders. Biallelic ELFN1 variants disrupt synaptic protein trafficking to the cell surface—validated through functional assays and mouse/zebrafish models.
Neurogenetics alert! Biallelic loss of function variants in ELFN1 cause a neurodevelopmental disorder with DD/ID, seizures and movement disorder. bit.ly/4lLrZ46

July 26, 2025 at 10:11 PM
Our new study characterises ELFN1 deficiency as a novel autosomal recessive neurodevelopmental disorder marked by epilepsy, GDD/ID, & movement disorders. Biallelic ELFN1 variants disrupt synaptic protein trafficking to the cell surface—validated through functional assays and mouse/zebrafish models.
We define the recessive ELOVL1-related disorder, part of an emerging group of neurocutaneous syndromes caused by biallelic ELOVL1/4 variants. Monoallelic ELOVL1/4/5 variants lead to spastic paraplegia & ataxia. These genes encode enzymes that elongate very-long-chain fatty acids. loom.ly/wwx-XQ4

Biallelic ELOVL1 Variants Are Linked to Hypomyelinating Leukodystrophy, Movement Disorder, and Ichthyosis
Background Very long chain fatty acids (VLCFAs) are an integral component of myelin and the epidermal water barrier. Variants in genes encoding enzymes responsible for catalyzing the first and rate ...
loom.ly
July 14, 2025 at 6:57 AM
We define the recessive ELOVL1-related disorder, part of an emerging group of neurocutaneous syndromes caused by biallelic ELOVL1/4 variants. Monoallelic ELOVL1/4/5 variants lead to spastic paraplegia & ataxia. These genes encode enzymes that elongate very-long-chain fatty acids. loom.ly/wwx-XQ4
Reposted by Reza Maroofian

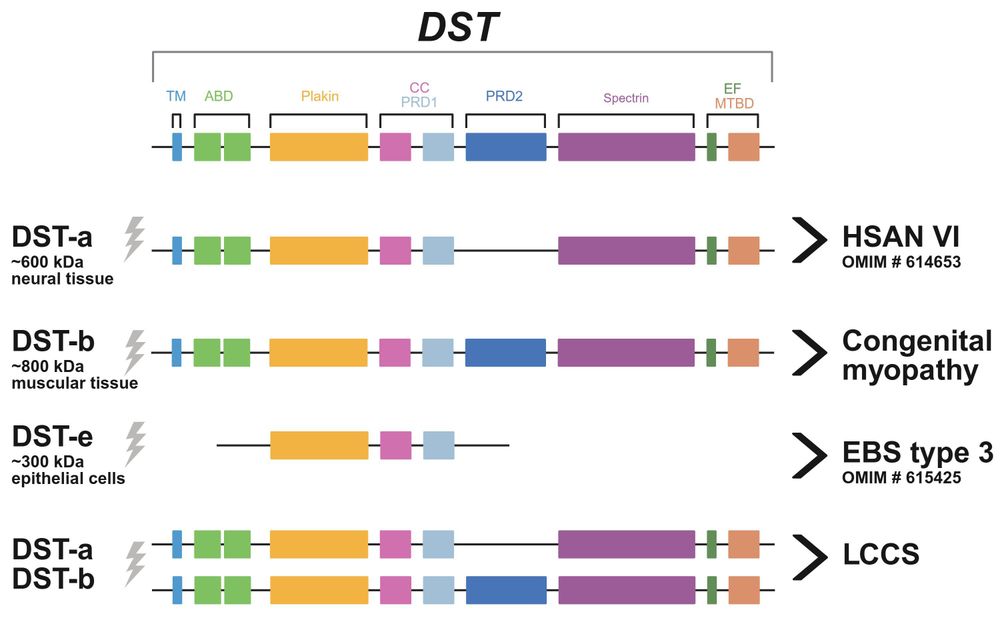
The dystonin gene encodes three major isoforms: DST-a, -b, and -e. Jacob et al. report that variants exclusively affecting DST-b cause an autosomal recessive congenital myopathy, while variants that also affect DST-a cause a lethal contracture syndrome. tinyurl.com/3p99pu9j
July 3, 2025 at 9:49 AM
The dystonin gene encodes three major isoforms: DST-a, -b, and -e. Jacob et al. report that variants exclusively affecting DST-b cause an autosomal recessive congenital myopathy, while variants that also affect DST-a cause a lethal contracture syndrome. tinyurl.com/3p99pu9j
We define the critical role of the LGI1–ADAM22/23 pathway in developmental & epileptic encephalopathy (DEE)—essential for regulating synaptic transmission and brain excitability. Previously linked ADAM22 to DEE, now we report ultra-rare biallelic LGI1 & ADAM23 variants causing lethal DEE.
Hirano et al. define a novel neurological disease spectrum involving the LGI1–ADAM22/23 pathway, identifying ultra-rare biallelic LGI1 variants in developmental and epileptic encephalopathy, and a biallelic ADAM23 variant in lethal neonatal epilepsy. tinyurl.com/4cpc48fy

June 18, 2025 at 6:00 PM
We define the critical role of the LGI1–ADAM22/23 pathway in developmental & epileptic encephalopathy (DEE)—essential for regulating synaptic transmission and brain excitability. Previously linked ADAM22 to DEE, now we report ultra-rare biallelic LGI1 & ADAM23 variants causing lethal DEE.
Loss of XRCC1 disrupts cerebellar development in zebrafish due to toxic PARP1 accumulation. Strikingly, parp1 knockdown rescues the XRCC1 phenotype, supporting PARP1 inhibition as a potential therapy in recessive XRCC1-related neurodegenerative disorders with ataxia. www.nature.com/articles/s41...

Parp1 deletion rescues cerebellar hypotrophy in xrcc1 mutant zebrafish - Scientific Reports
Scientific Reports - Parp1 deletion rescues cerebellar hypotrophy in xrcc1 mutant zebrafish
www.nature.com
May 18, 2025 at 4:23 PM
Loss of XRCC1 disrupts cerebellar development in zebrafish due to toxic PARP1 accumulation. Strikingly, parp1 knockdown rescues the XRCC1 phenotype, supporting PARP1 inhibition as a potential therapy in recessive XRCC1-related neurodegenerative disorders with ataxia. www.nature.com/articles/s41...
Reposted by Reza Maroofian

Proud to present our work identifying a role for DIAPH1 and gamma-actin in regulating DSB repair and how defects in this pathway give rise to human disease. Big thanks to everyone involved, especially Beth Woodward, Sudipta Lahiri and @anoopsinghchauhan.bsky.social. www.nature.com/articles/s41...

Inherited deficiency of DIAPH1 identifies a DNA double strand break repair pathway regulated by γ-actin - Nature Communications
DNA double strand break repair pathways ensure genome stability and prevent disease. Here the authors show that the actin nucleating factor DIAPH1 and γ-actin promote homologous recombination (HR)-dep...
www.nature.com
May 14, 2025 at 7:59 PM
Proud to present our work identifying a role for DIAPH1 and gamma-actin in regulating DSB repair and how defects in this pathway give rise to human disease. Big thanks to everyone involved, especially Beth Woodward, Sudipta Lahiri and @anoopsinghchauhan.bsky.social. www.nature.com/articles/s41...

We previously reported a novel recessive pediatric neurodegenerative disorder linked to BORCS8. Now, we identify another BORC complex subunit, BORCS5, as a new disease gene causing a broader neurodevelopmental & neurodegenerative spectrum with clear genotype–phenotype correlation. Read the preprint:

Pathogenic variants in BORCS5 Cause a Spectrum of Neurodevelopmental and Neurodegenerative Disorders with Lysosomal Dysfunction https://www.medrxiv.org/content/10.1101/2025.04.30.25326597v1
May 8, 2025 at 10:18 PM
We previously reported a novel recessive pediatric neurodegenerative disorder linked to BORCS8. Now, we identify another BORC complex subunit, BORCS5, as a new disease gene causing a broader neurodevelopmental & neurodegenerative spectrum with clear genotype–phenotype correlation. Read the preprint:
Reposted by Reza Maroofian
📣New from @rdexeter.bsky.social & co!
📄Bi-allelic UGGT1 variants cause a congenital disorder of glycosylation
📄Bi-allelic UGGT1 variants cause a congenital disorder of glycosylation
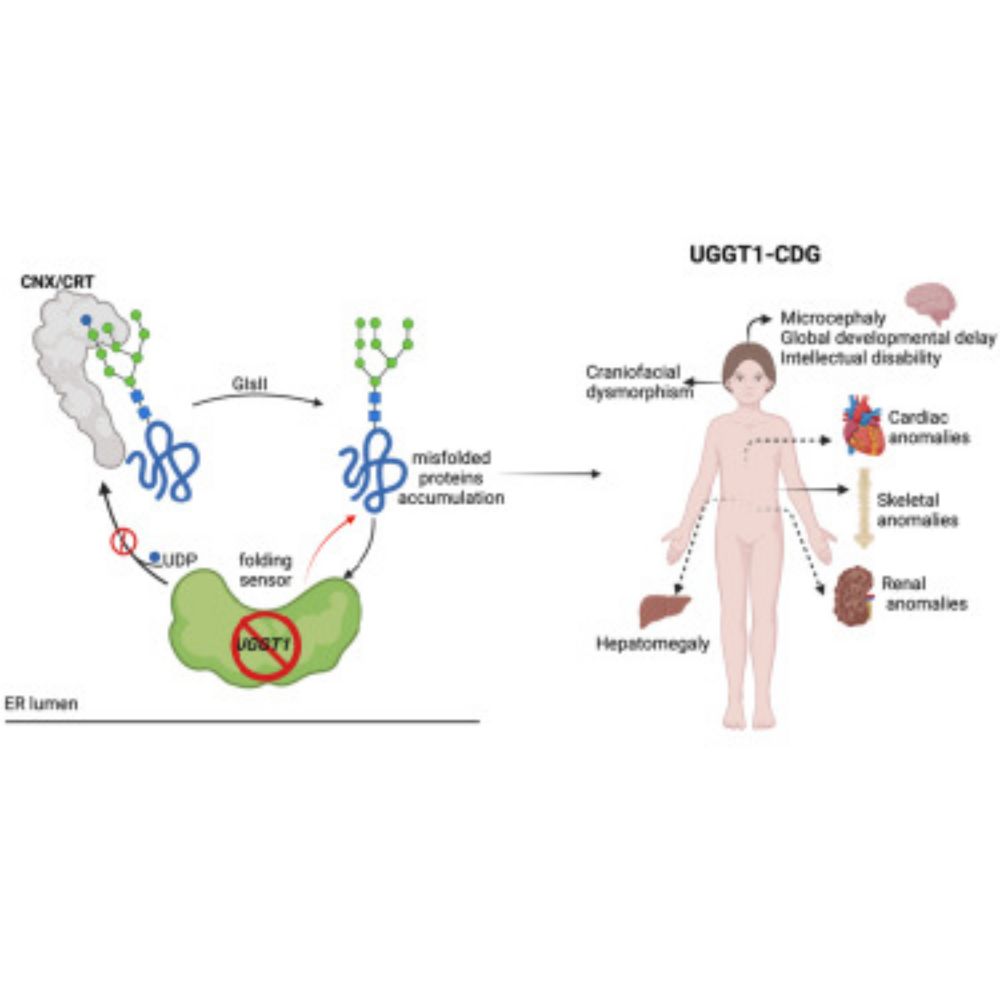
Bi-allelic UGGT1 variants cause a congenital disorder of glycosylation
Bi-allelic UGGT1 variants cause a distinct congenital disorder of glycosylation (UGGT1-CDG)
with variable severity, characterized by neurodevelopmental impairment, seizures,
dysmorphic features, and m...
www.cell.com
April 22, 2025 at 4:49 PM
📣New from @rdexeter.bsky.social & co!
📄Bi-allelic UGGT1 variants cause a congenital disorder of glycosylation
📄Bi-allelic UGGT1 variants cause a congenital disorder of glycosylation
Our lab characterises the autosomal recessive TRMT1-related neurodevelopmental disorder through a large cohort, patient-derived cells, and zebrafish model—linking defective tRNA methylation to intellectual disability and expanding the emerging group of "tRNAopathies". www.cell.com/ajhg/fulltex...

Biallelic pathogenic variants in TRMT1 disrupt tRNA modification and induce a neurodevelopmental disorder
We identify bi-allelic variants in TRMT1, encoding a tRNA-modification enzyme, that
cause intellectual disability and developmental delay. Functional studies in human
cells and zebrafish provide insig...
www.cell.com
April 19, 2025 at 12:33 PM
Our lab characterises the autosomal recessive TRMT1-related neurodevelopmental disorder through a large cohort, patient-derived cells, and zebrafish model—linking defective tRNA methylation to intellectual disability and expanding the emerging group of "tRNAopathies". www.cell.com/ajhg/fulltex...
Reposted by Reza Maroofian
📣Online now!
📄Bi-allelic variants in MRPL49 cause variable clinical presentations, including sensorineural hearing loss, leukodystrophy, and ovarian insufficiency
📄Bi-allelic variants in MRPL49 cause variable clinical presentations, including sensorineural hearing loss, leukodystrophy, and ovarian insufficiency

Bi-allelic variants in MRPL49 cause variable clinical presentations, including sensorineural hearing loss, leukodystrophy, and ovarian insufficiency
We identify bi-allelic variants in the mitoribosomal large subunit encoded by MRPL49
as a cause of a pleiotropic presentation of hearing loss, ovarian failure, learning
disability, and leukodystrophy ...
www.cell.com
March 5, 2025 at 3:58 PM
📣Online now!
📄Bi-allelic variants in MRPL49 cause variable clinical presentations, including sensorineural hearing loss, leukodystrophy, and ovarian insufficiency
📄Bi-allelic variants in MRPL49 cause variable clinical presentations, including sensorineural hearing loss, leukodystrophy, and ovarian insufficiency
Reposted by Reza Maroofian

For #RareDiseaseDay2025, @qs-neurogenetics.bsky.social is celebrating the invaluable contributions of our international collaborators, whose dedication is driving ground-breaking advancements in rare disease research across the globe. ucl.ac.uk/ion/news/202...

February 28, 2025 at 9:27 AM
For #RareDiseaseDay2025, @qs-neurogenetics.bsky.social is celebrating the invaluable contributions of our international collaborators, whose dedication is driving ground-breaking advancements in rare disease research across the globe. ucl.ac.uk/ion/news/202...
Reposted by Reza Maroofian

Prof. Shamima Rahman comments on ‘Biallelic NDUFA13 variants lead to a neurodevelopmental phenotype with gradual neurological impairment’ by Kaiyrzhanov et al, highlighting NDUFA13 deficiency in Leigh syndrome, linking neuroinflammation & imaging. Please read at: buff.ly/3D5Dtyf
February 21, 2025 at 11:01 AM
Prof. Shamima Rahman comments on ‘Biallelic NDUFA13 variants lead to a neurodevelopmental phenotype with gradual neurological impairment’ by Kaiyrzhanov et al, highlighting NDUFA13 deficiency in Leigh syndrome, linking neuroinflammation & imaging. Please read at: buff.ly/3D5Dtyf
As part of 2 parallel studies, we delineated a new subtype of neurodevelopmental disorder linked to biallelic GTF3C3 variants. One study models the disorder using zebrafish, while the other utilizes fly. Check both papers below: academic.oup.com/braincomms/a... www.sciencedirect.com/science/arti...

Biallelic variants in GTF3C3 encoding a subunit of the TFIIIC2 complex are associated with neurodevelopmental phenotypes in humans and zebrafish
Abdel-Hamid et al. identified biallelic GTF3C3 variants in four individuals with neurodevelopmental disorders, including developmental delay/intellectual d
academic.oup.com
February 5, 2025 at 7:22 PM
As part of 2 parallel studies, we delineated a new subtype of neurodevelopmental disorder linked to biallelic GTF3C3 variants. One study models the disorder using zebrafish, while the other utilizes fly. Check both papers below: academic.oup.com/braincomms/a... www.sciencedirect.com/science/arti...
NDUFA13, a mitochondrial complex I subunit, was linked to complex I deficiency in only 3 patients. We now report 10 more cases, expanding the phenotypic spectrum, consolidating its role, & comparing it with other complex I deficiency subtypes. Please Check our paper: academic.oup.com/braincomms/a...

Biallelic NDUFA13 variants lead to a neurodevelopmental phenotype with gradual neurological impairment
Kaiyrzhanov et al. provide a cumulative phenotype characterization of NADH-ubiquinone oxidoreductase 1 alpha subcomplex 13 (NDUFA13)-related disease descri
academic.oup.com
January 24, 2025 at 11:55 PM
NDUFA13, a mitochondrial complex I subunit, was linked to complex I deficiency in only 3 patients. We now report 10 more cases, expanding the phenotypic spectrum, consolidating its role, & comparing it with other complex I deficiency subtypes. Please Check our paper: academic.oup.com/braincomms/a...
In 2013, we identified KPTN as a cause of NDD, though its function was unclear at the time. By 2017, it was shown to be part of the KICSTOR complex with KICS2, ITFG2, & SZT2, regulating mTORC1 signaling. We've now published KICS2 linked to NDD, with ITFG2 next in line. www.cell.com/ajhg/fulltex...

Bi-allelic KICS2 mutations impair KICSTOR complex-mediated mTORC1 regulation, causing intellectual disability and epilepsy
Sequencing 8 individuals with intellectual disability identified bi-allelic variants
in KICS2, which encodes a component of the KICSTOR complex. A combination of in vitro
and in vivo analyses demonstr...
www.cell.com
January 16, 2025 at 5:48 PM
In 2013, we identified KPTN as a cause of NDD, though its function was unclear at the time. By 2017, it was shown to be part of the KICSTOR complex with KICS2, ITFG2, & SZT2, regulating mTORC1 signaling. We've now published KICS2 linked to NDD, with ITFG2 next in line. www.cell.com/ajhg/fulltex...
TRMT1 & TRMT1L modify tRNAs, essential for protein production. Their modifications are crucial for tRNA stability & function. Biallelic TRMT1 variants are linked to intellectual disability, while TRMT1L variants lead to a neurodegenerative disorder. Check our new paper! www.cell.com/cell-reports...

Human TRMT1 and TRMT1L paralogs ensure the proper modification state, stability, and function of tRNAs
Zhang et al. elucidate the targets of a tRNA modification enzyme family and identify unanticipated roles for a duplicated tRNA modification enzyme that can be uncoupled from its ancestral function. These findings uncover the molecular mechanisms by which tRNAs are dysregulated in human disorders caused by tRNA modification deficiency.
www.cell.com
January 11, 2025 at 12:53 AM
TRMT1 & TRMT1L modify tRNAs, essential for protein production. Their modifications are crucial for tRNA stability & function. Biallelic TRMT1 variants are linked to intellectual disability, while TRMT1L variants lead to a neurodegenerative disorder. Check our new paper! www.cell.com/cell-reports...
We report 35 patients with biallelic RBL2 loss-of-function variants presenting with developmental delay/intellectual disability, hypotonia, seizures, microcephaly & brain abnormalities. Drosophila models recapitulate key features & suggest RBL2 re-expression may help rescue neurological symptoms.
Very happy to see our work with Henry Houlden's group and others on RBL2/Rbf mutations linked to a multifaceted neurodevelopmental disorder, published in Brain today. This work was led by super post-doc @gabrielaughey.bsky.social, Elisa Cali, and Reza Maroofian.
academic.oup.com/brain/advanc...
academic.oup.com/brain/advanc...

Clinical and genetic characterization of a progressive RBL2-associated neurodevelopmental disorder
RBL2 dysfunction, which disrupts cell-cycle gene expression, has been linked to a severe neurodevelopmental disorder. Aughey et al. characterize a cohort o
academic.oup.com
January 6, 2025 at 6:57 PM
We report 35 patients with biallelic RBL2 loss-of-function variants presenting with developmental delay/intellectual disability, hypotonia, seizures, microcephaly & brain abnormalities. Drosophila models recapitulate key features & suggest RBL2 re-expression may help rescue neurological symptoms.
In 2021, we identified a rare VWA1 founder mutation in UK & Western European populations, linked to neuromuscular disorder but elusive due to low genomic coverage. We now report an expanded phenotypic spectrum in a global cohort with this recurrent mutation & other VWA1 variants.
Nagy et al. report that VWA1-related motor neuropathy is linked to 11 novel mutations and diverse symptoms, including hypermobility and upper motor neuron signs. This highlights the importance of understanding genetic and phenotypic variability to improve diagnosis. Read at: https://buff.ly/3ZZy57v

January 5, 2025 at 12:45 PM
In 2021, we identified a rare VWA1 founder mutation in UK & Western European populations, linked to neuromuscular disorder but elusive due to low genomic coverage. We now report an expanded phenotypic spectrum in a global cohort with this recurrent mutation & other VWA1 variants.