



Evan Qu
@quevan.bsky.social
PhD candidate in the MIT Microbiology Program (co2020). @Lieberman Lab @contaminatedsci.bsky.social. I study the ecology and evolution of skin microbes, like this one -> 🤏
PHLAME is available to try out on GitHub:
github.com/quevan/phlame
github.com/quevan/phlame
GitHub - quevan/phlame
Contribute to quevan/phlame development by creating an account on GitHub.
github.com
February 11, 2025 at 11:06 PM
PHLAME is available to try out on GitHub:
github.com/quevan/phlame
github.com/quevan/phlame
Turning to the vaginal microbiome, we showed how PHLAME's novelty aware approach can identify samples that are abundant novel diversity.
By our estimate, about 1/3 of vaginal samples we looked at had substantial abundances (>20%) of yet-characterized Gardnerella strains
By our estimate, about 1/3 of vaginal samples we looked at had substantial abundances (>20%) of yet-characterized Gardnerella strains
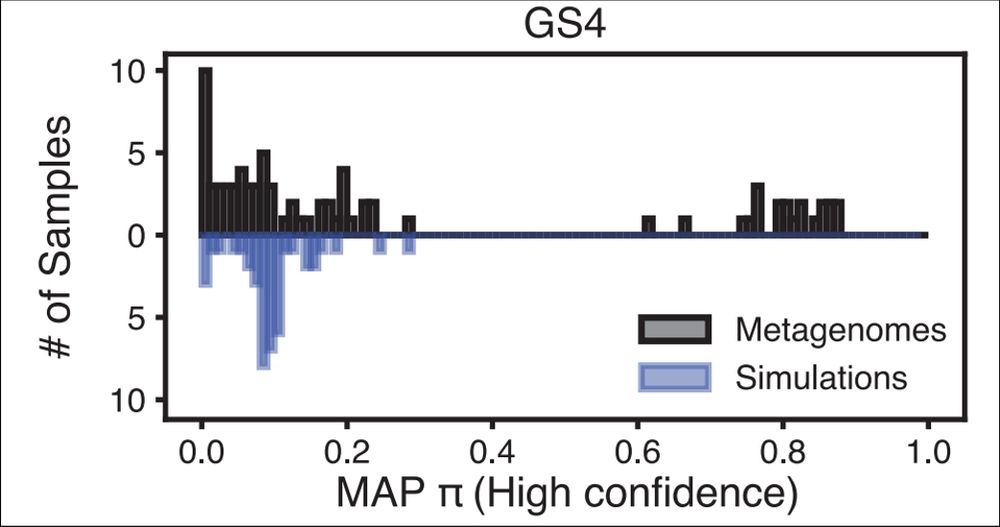
February 11, 2025 at 11:06 PM
Turning to the vaginal microbiome, we showed how PHLAME's novelty aware approach can identify samples that are abundant novel diversity.
By our estimate, about 1/3 of vaginal samples we looked at had substantial abundances (>20%) of yet-characterized Gardnerella strains
By our estimate, about 1/3 of vaginal samples we looked at had substantial abundances (>20%) of yet-characterized Gardnerella strains
We also found a clade of C. acnes that is higher abundance on older people (>40 yo). This association is independent of sex and consistent across geographic regions.

February 11, 2025 at 11:06 PM
We also found a clade of C. acnes that is higher abundance on older people (>40 yo). This association is independent of sex and consistent across geographic regions.
We used PHLAME to pull out some interesting associations from public data.
In the skin microbiome, we discovered that some clades of C. acnes are recently emerged, strongly geographic restricted, and at high prevalence in those regions. This pattern may indicate region specific adaptation.
In the skin microbiome, we discovered that some clades of C. acnes are recently emerged, strongly geographic restricted, and at high prevalence in those regions. This pattern may indicate region specific adaptation.

February 11, 2025 at 11:06 PM
We used PHLAME to pull out some interesting associations from public data.
In the skin microbiome, we discovered that some clades of C. acnes are recently emerged, strongly geographic restricted, and at high prevalence in those regions. This pattern may indicate region specific adaptation.
In the skin microbiome, we discovered that some clades of C. acnes are recently emerged, strongly geographic restricted, and at high prevalence in those regions. This pattern may indicate region specific adaptation.
Using this novelty-aware approach, PHLAME achieves near-perfect precision and high sensitivity, even for species with low coverage.
We also benchmarked PHLAME using @microjacob.bsky.social's unique resource of thousands of paired isolates and metagenomes from the same samples (see Fig. 4)
We also benchmarked PHLAME using @microjacob.bsky.social's unique resource of thousands of paired isolates and metagenomes from the same samples (see Fig. 4)

February 11, 2025 at 11:06 PM
Using this novelty-aware approach, PHLAME achieves near-perfect precision and high sensitivity, even for species with low coverage.
We also benchmarked PHLAME using @microjacob.bsky.social's unique resource of thousands of paired isolates and metagenomes from the same samples (see Fig. 4)
We also benchmarked PHLAME using @microjacob.bsky.social's unique resource of thousands of paired isolates and metagenomes from the same samples (see Fig. 4)
Counting missing mutations is not easy when reads are low-coverage and overdispersed.
We solved this problem using a model that independently measures dispersion and zero-inflation by comparing counts across just the mutational allele compared to all alleles at the same positions.
We solved this problem using a model that independently measures dispersion and zero-inflation by comparing counts across just the mutational allele compared to all alleles at the same positions.

February 11, 2025 at 11:06 PM
Counting missing mutations is not easy when reads are low-coverage and overdispersed.
We solved this problem using a model that independently measures dispersion and zero-inflation by comparing counts across just the mutational allele compared to all alleles at the same positions.
We solved this problem using a model that independently measures dispersion and zero-inflation by comparing counts across just the mutational allele compared to all alleles at the same positions.
We figured out that we could estimate the divergence along each branch (novelty of a new strain) by counting the proportion of clade-specific mutations missing in metagenomic samples (π).

February 11, 2025 at 11:06 PM
We figured out that we could estimate the divergence along each branch (novelty of a new strain) by counting the proportion of clade-specific mutations missing in metagenomic samples (π).
PHLAME quantifies novel strain diversity in samples using an evolutionary framework. Novel strains in a sample are assumed to share some, but not all, evolutionary history with known strains. We call the degree of unshared evolutionary history between a sample and a reference database Divergence.

February 11, 2025 at 11:06 PM
PHLAME quantifies novel strain diversity in samples using an evolutionary framework. Novel strains in a sample are assumed to share some, but not all, evolutionary history with known strains. We call the degree of unshared evolutionary history between a sample and a reference database Divergence.
We were concerned that ‘pushing’ novel diversity might influence downstream association detection, especially in samples with significant amounts of novel strain diversity.

February 11, 2025 at 11:06 PM
We were concerned that ‘pushing’ novel diversity might influence downstream association detection, especially in samples with significant amounts of novel strain diversity.
Standard practice for complex metagenomic samples is to use reference databases to detect strains.
Because reference databases are never comprehensive, many of these methods will represent novel strains (i.e., not in the database) as a nearby representative genome in the database.
Because reference databases are never comprehensive, many of these methods will represent novel strains (i.e., not in the database) as a nearby representative genome in the database.
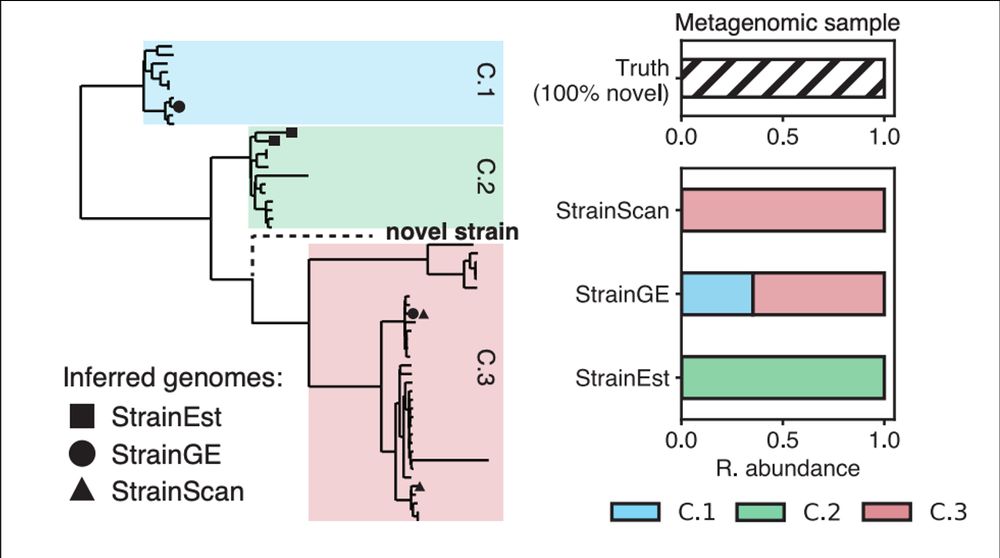
February 11, 2025 at 11:06 PM
Standard practice for complex metagenomic samples is to use reference databases to detect strains.
Because reference databases are never comprehensive, many of these methods will represent novel strains (i.e., not in the database) as a nearby representative genome in the database.
Because reference databases are never comprehensive, many of these methods will represent novel strains (i.e., not in the database) as a nearby representative genome in the database.
Many health or environmental associations may be driven by intraspecies variants.
The most straightforward approach for strain associations, direct inference of genotypes from metagenomics, is difficult in environments where many strains of the same species coexist.
The most straightforward approach for strain associations, direct inference of genotypes from metagenomics, is difficult in environments where many strains of the same species coexist.
February 11, 2025 at 11:06 PM
Many health or environmental associations may be driven by intraspecies variants.
The most straightforward approach for strain associations, direct inference of genotypes from metagenomics, is difficult in environments where many strains of the same species coexist.
The most straightforward approach for strain associations, direct inference of genotypes from metagenomics, is difficult in environments where many strains of the same species coexist.