



GAMA Miguel Angel
@miangoar.bsky.social
Biologist that navigate in the oceans of diversity through space-time
Protein evolution, metagenomics, AI/ML/DL
Website https://miangoaren.github.io/
Protein evolution, metagenomics, AI/ML/DL
Website https://miangoaren.github.io/
I strongly recommend making cat-based diagrams to illustrate complex topics in protein science: "Figure 4 considers [...] invariance and equivariance with respect to translations and rotations in 3D. For illustration purposes, the figure includes a series of cat cartoons in 2D."

October 24, 2025 at 5:47 AM
I strongly recommend making cat-based diagrams to illustrate complex topics in protein science: "Figure 4 considers [...] invariance and equivariance with respect to translations and rotations in 3D. For illustration purposes, the figure includes a series of cat cartoons in 2D."
I just want to create hype and say that I made a 10-class course to introduce people to AI-driven protein design. It’s around 750 slides and will be freely available for anyone who wants to use them and, most importantly, improve them. Stay tuned :)

October 9, 2025 at 5:59 PM
I just want to create hype and say that I made a 10-class course to introduce people to AI-driven protein design. It’s around 750 slides and will be freely available for anyone who wants to use them and, most importantly, improve them. Stay tuned :)
This is a breakthrough for protein science🔥AFAIK this is the largest protein DB, with >100B seqs (3B clustered at 50%). New biology will come from LOGAN: new folds, topologies, etc. You can also improve your AlphaFold models by building better MSAs. Future AI models will also use LOGAN for training

September 5, 2025 at 4:51 PM
This is a breakthrough for protein science🔥AFAIK this is the largest protein DB, with >100B seqs (3B clustered at 50%). New biology will come from LOGAN: new folds, topologies, etc. You can also improve your AlphaFold models by building better MSAs. Future AI models will also use LOGAN for training
12/13 Bindcraft started as a binder design tutorial for the Boston Protein Design and Modeling Club, and it evolved into one of the most promising tools in AI-based protein design. And Importantly, it is open-source!🤗
Congrats to all the authors!
Congrats to all the authors!
August 27, 2025 at 7:54 PM
12/13 Bindcraft started as a binder design tutorial for the Boston Protein Design and Modeling Club, and it evolved into one of the most promising tools in AI-based protein design. And Importantly, it is open-source!🤗
Congrats to all the authors!
Congrats to all the authors!
11/13 The authors have gone a step further and are currently developing BoltzDesign1, which instead of designing binders, focuses on biomolecular interactions between proteins and small molecules. However, one of the main limitations of both AIs is their high computational cost.

August 27, 2025 at 7:54 PM
11/13 The authors have gone a step further and are currently developing BoltzDesign1, which instead of designing binders, focuses on biomolecular interactions between proteins and small molecules. However, one of the main limitations of both AIs is their high computational cost.
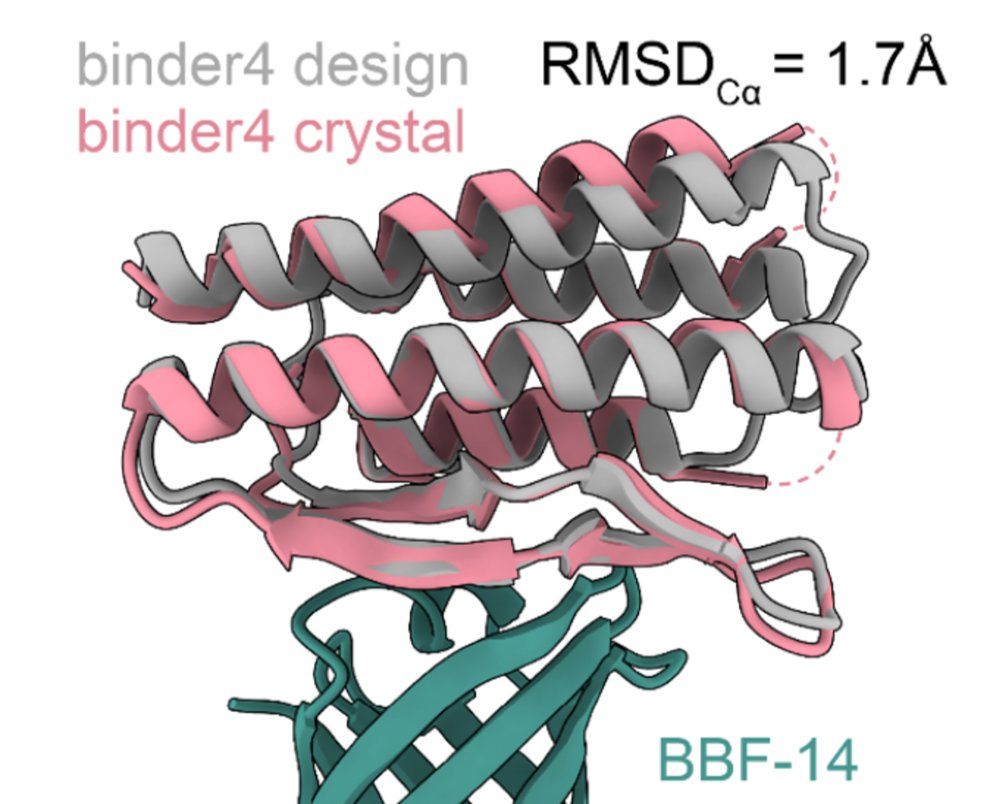
9/13 the most important results IMO was the determination of atomic structures of four binders, where in all cases, the computational designs were highly consistent with the experimentally determined ones.
August 27, 2025 at 7:54 PM
9/13 the most important results IMO was the determination of atomic structures of four binders, where in all cases, the computational designs were highly consistent with the experimentally determined ones.
8/13 They designed binders targeting:
*proteins with no known binding sites
*membrane proteins , which are much harder than intra/extra-cellular proteins
*proteins lacking evolutionary information
*proteins that interact with DNA/RNA
*medically relevant proteins such as those causing allergies
*proteins with no known binding sites
*membrane proteins , which are much harder than intra/extra-cellular proteins
*proteins lacking evolutionary information
*proteins that interact with DNA/RNA
*medically relevant proteins such as those causing allergies

August 27, 2025 at 7:54 PM
8/13 They designed binders targeting:
*proteins with no known binding sites
*membrane proteins , which are much harder than intra/extra-cellular proteins
*proteins lacking evolutionary information
*proteins that interact with DNA/RNA
*medically relevant proteins such as those causing allergies
*proteins with no known binding sites
*membrane proteins , which are much harder than intra/extra-cellular proteins
*proteins lacking evolutionary information
*proteins that interact with DNA/RNA
*medically relevant proteins such as those causing allergies
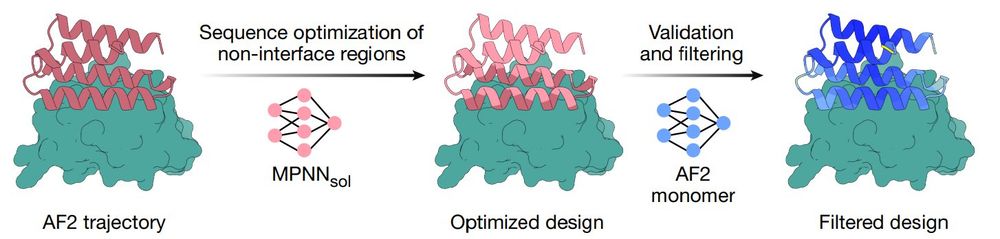
7/13 Then it uses ProteinMPNN to optimize for solubility, increasing the chances of experimental success. Finally, uses AF2 to predict the structure. To demonstrate Bindcraft’s utility, the authors carried out many wet-lab experiments, something not as common as I would like.
August 27, 2025 at 7:54 PM
7/13 Then it uses ProteinMPNN to optimize for solubility, increasing the chances of experimental success. Finally, uses AF2 to predict the structure. To demonstrate Bindcraft’s utility, the authors carried out many wet-lab experiments, something not as common as I would like.
6/13 Bindcraft takes advantage of this by first proposing a random seq and predicting its structure to assess how well it interacts with the target protein. It then uses info from each interaction, successful or not, to optimize the seqs until it arrives at a credible interaction

August 27, 2025 at 7:54 PM
6/13 Bindcraft takes advantage of this by first proposing a random seq and predicting its structure to assess how well it interacts with the target protein. It then uses info from each interaction, successful or not, to optimize the seqs until it arrives at a credible interaction
5/13 Bindcraft is an improved version of AlphaFold2, specifically AF-Multimer, which predicts the structure of protein complexes. Having been trained on thousands of structures, AF-Multimer learned to identify which sites are most likely to form protein–protein interactions.

August 27, 2025 at 7:54 PM
5/13 Bindcraft is an improved version of AlphaFold2, specifically AF-Multimer, which predicts the structure of protein complexes. Having been trained on thousands of structures, AF-Multimer learned to identify which sites are most likely to form protein–protein interactions.
4/13 Bindcraft designs both the sequence and structure of binders, achieving a success rate between 10-100%, since designing large or complex binders is more challenging. This is enormous, considering that our previous best physics/biochemistry-based methods reached a 0.1%.

August 27, 2025 at 7:54 PM
4/13 Bindcraft designs both the sequence and structure of binders, achieving a success rate between 10-100%, since designing large or complex binders is more challenging. This is enormous, considering that our previous best physics/biochemistry-based methods reached a 0.1%.
3/13 We have learned how to design PPI so that one protein, called a binder, can bind to another and regulate it. e.g., cancer drugs are binders. However, designing binders requires yrs of research and detailed biomolecular knowledge. So, what if we teach an AI to design binders?

August 27, 2025 at 7:54 PM
3/13 We have learned how to design PPI so that one protein, called a binder, can bind to another and regulate it. e.g., cancer drugs are binders. However, designing binders requires yrs of research and detailed biomolecular knowledge. So, what if we teach an AI to design binders?
2/13 Proteins carry out many functions on their own, but when they interact with each other, they generate a diversity of mechanisms that expand and regulate those functions. PPI arose over millions of years of evolution, giving rise to processes as complex as metabolism.

August 27, 2025 at 7:54 PM
2/13 Proteins carry out many functions on their own, but when they interact with each other, they generate a diversity of mechanisms that expand and regulate those functions. PPI arose over millions of years of evolution, giving rise to processes as complex as metabolism.
1/13 🧵 Today, Bindcraft was published in
@nature.com , one of the most famous AIs in biology for designing protein–protein interactions (PPI). In my opinion. Bindcraft represents one of the most important advances in the post–AlphaFold2 era.
@nature.com , one of the most famous AIs in biology for designing protein–protein interactions (PPI). In my opinion. Bindcraft represents one of the most important advances in the post–AlphaFold2 era.

August 27, 2025 at 7:54 PM
1/13 🧵 Today, Bindcraft was published in
@nature.com , one of the most famous AIs in biology for designing protein–protein interactions (PPI). In my opinion. Bindcraft represents one of the most important advances in the post–AlphaFold2 era.
@nature.com , one of the most famous AIs in biology for designing protein–protein interactions (PPI). In my opinion. Bindcraft represents one of the most important advances in the post–AlphaFold2 era.
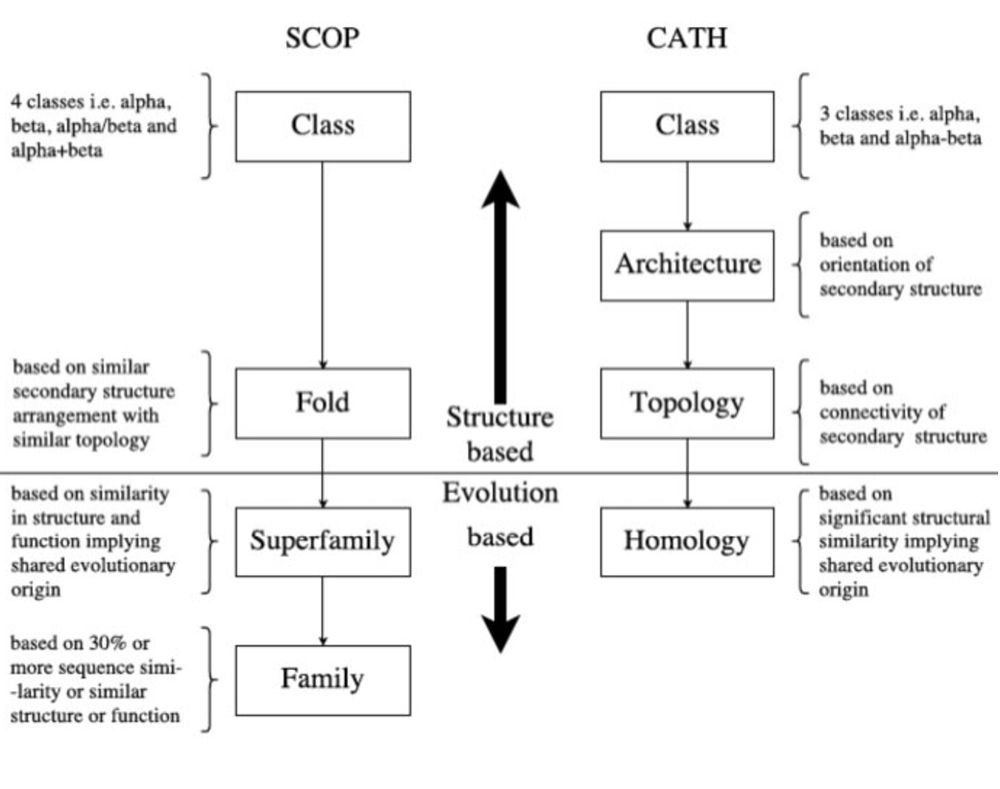
Does anyone know of a recent comparison of the main structural classification schemes of proteins and guidance on when to choose one? Something like this but including ECOD and perhaps seq-based schemes like Pfam, SUPERFAMILY and CDD.
Img source (2020)
pubmed.ncbi.nlm.nih.gov/32302382/
Img source (2020)
pubmed.ncbi.nlm.nih.gov/32302382/
August 27, 2025 at 2:16 AM
Does anyone know of a recent comparison of the main structural classification schemes of proteins and guidance on when to choose one? Something like this but including ECOD and perhaps seq-based schemes like Pfam, SUPERFAMILY and CDD.
Img source (2020)
pubmed.ncbi.nlm.nih.gov/32302382/
Img source (2020)
pubmed.ncbi.nlm.nih.gov/32302382/
I think that this pic from the original paper illustrates very well how fast Diamond2 is. I'm also waiting for the final publication of the Diamond Deepclust DB that is the largest protein seq DB AFAIK
Sensitive protein alignments at tree-of-life scale using DIAMOND
www.nature.com/articles/s41...
Sensitive protein alignments at tree-of-life scale using DIAMOND
www.nature.com/articles/s41...

July 21, 2025 at 5:24 PM
I think that this pic from the original paper illustrates very well how fast Diamond2 is. I'm also waiting for the final publication of the Diamond Deepclust DB that is the largest protein seq DB AFAIK
Sensitive protein alignments at tree-of-life scale using DIAMOND
www.nature.com/articles/s41...
Sensitive protein alignments at tree-of-life scale using DIAMOND
www.nature.com/articles/s41...
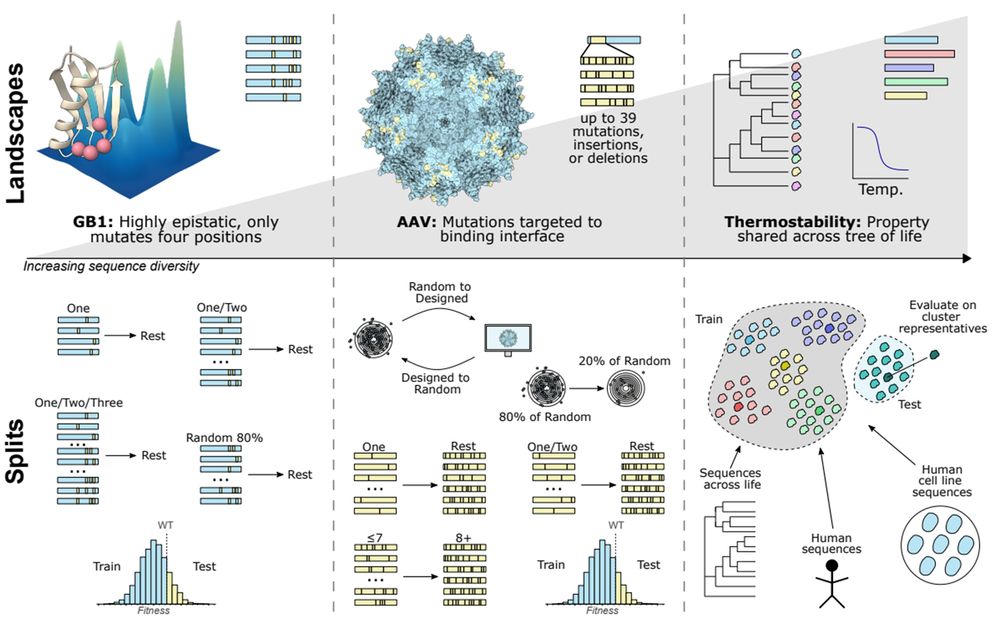
9/9 Perhaps the ~1.6k de novo designs collected by The PDA should be a good choice to evaluate further models, which also need to take into consideration ways to prevent data leakage. OOD appears to be harder than ID generalization and each task requieres a particular split
bsky.app/profile/bris...
bsky.app/profile/bris...
July 9, 2025 at 5:36 PM
9/9 Perhaps the ~1.6k de novo designs collected by The PDA should be a good choice to evaluate further models, which also need to take into consideration ways to prevent data leakage. OOD appears to be harder than ID generalization and each task requieres a particular split
bsky.app/profile/bris...
bsky.app/profile/bris...
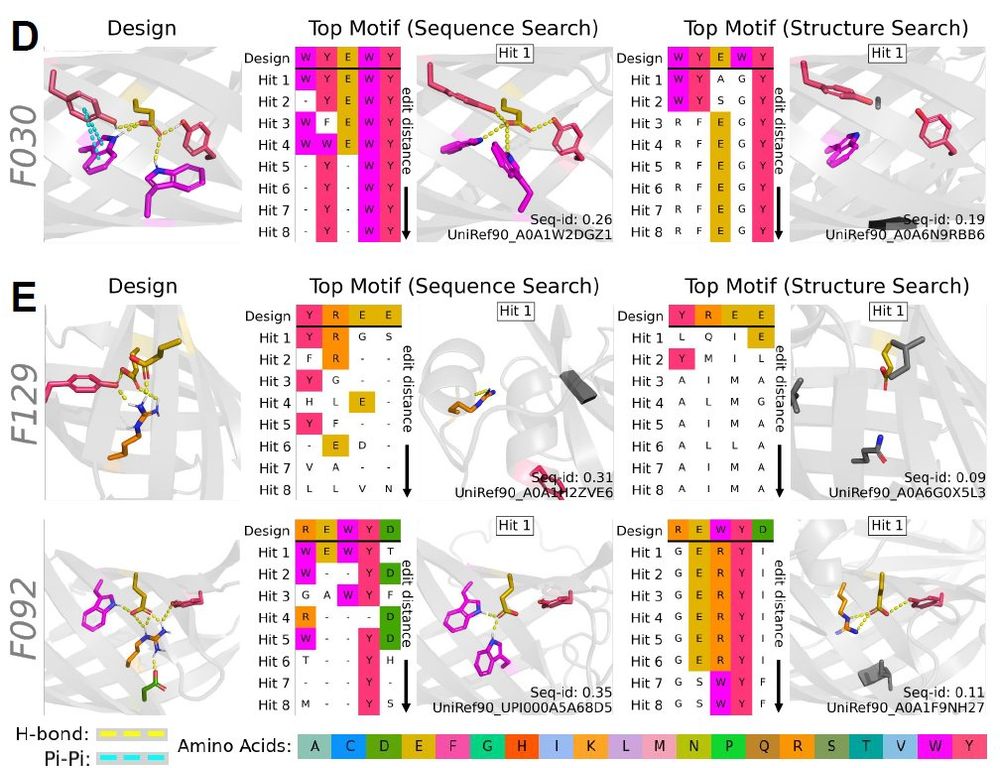
8/9 This is the only paper AFAIK that shows experimental validation for generated seqs below a 30% seqID threshold. They also demonstrate how their protocol based on ESM2 generates proteins with motifs surrounded by different contexts (D), as well as completely new motifs (E).
July 9, 2025 at 5:36 PM
8/9 This is the only paper AFAIK that shows experimental validation for generated seqs below a 30% seqID threshold. They also demonstrate how their protocol based on ESM2 generates proteins with motifs surrounded by different contexts (D), as well as completely new motifs (E).
7/9 So, what does OOD generalization mean? I don't know, but perhaps the answer lies in de novo proteins. In this paper, they applied fixed-backbone generation for various de novo designs and experimentally validated them, and their distribution is different from natural ones.

July 9, 2025 at 5:36 PM
7/9 So, what does OOD generalization mean? I don't know, but perhaps the answer lies in de novo proteins. In this paper, they applied fixed-backbone generation for various de novo designs and experimentally validated them, and their distribution is different from natural ones.
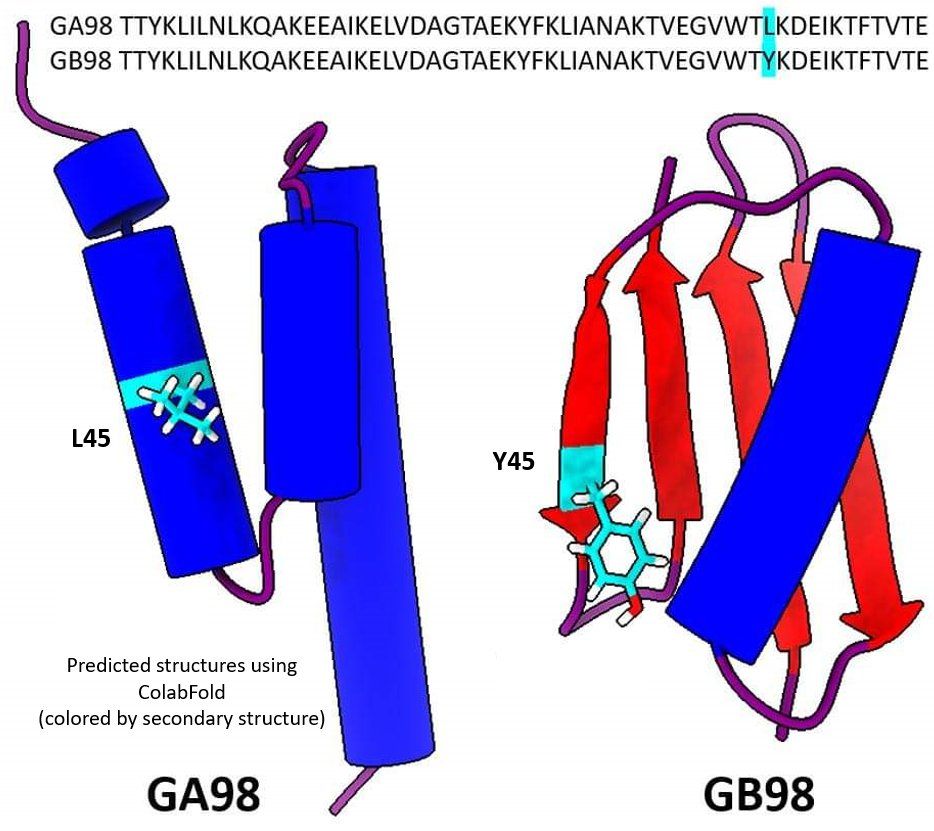
6/9 So, the problem seems to be the seqs. What if we split by structures? Perhaps it gets better, but there are still issues like convergence (e.g. pockets of serinproteases or carbonic anhydrases) and fold switching (estimated to occur in ~4% of the PDB)

July 9, 2025 at 5:36 PM
6/9 So, the problem seems to be the seqs. What if we split by structures? Perhaps it gets better, but there are still issues like convergence (e.g. pockets of serinproteases or carbonic anhydrases) and fold switching (estimated to occur in ~4% of the PDB)
5/9 Data leakage is not the only problem. We have others such as:
1 historical contingency (i.e., today’s proteins are not truly representative of the full diversity).
2 Proteins are not IID (due to factors like superfolds and shared motifs between non-homologous proteins)
1 historical contingency (i.e., today’s proteins are not truly representative of the full diversity).
2 Proteins are not IID (due to factors like superfolds and shared motifs between non-homologous proteins)

July 9, 2025 at 5:36 PM
5/9 Data leakage is not the only problem. We have others such as:
1 historical contingency (i.e., today’s proteins are not truly representative of the full diversity).
2 Proteins are not IID (due to factors like superfolds and shared motifs between non-homologous proteins)
1 historical contingency (i.e., today’s proteins are not truly representative of the full diversity).
2 Proteins are not IID (due to factors like superfolds and shared motifs between non-homologous proteins)
4/9 However, the commonly used seqID thresholds lie in the twilight zone (between 25-30% bsky.app/profile/mian...). This means that not all homologous seqs are divided correctly, leading to data leakage. e.g. Although these betalactamases share 12% seqID, they are homologous.

July 9, 2025 at 5:36 PM
4/9 However, the commonly used seqID thresholds lie in the twilight zone (between 25-30% bsky.app/profile/mian...). This means that not all homologous seqs are divided correctly, leading to data leakage. e.g. Although these betalactamases share 12% seqID, they are homologous.
3/9 When training a model, it’s common to create train/test splits by clustering sequences at a certain identity threshold (e.g. 30–50% in ESM2 or ProGen2). Then, train on one split and test on the split with seqs with low seqID. Ideally, these seqs should not be homologous, IMO.

July 9, 2025 at 5:36 PM
3/9 When training a model, it’s common to create train/test splits by clustering sequences at a certain identity threshold (e.g. 30–50% in ESM2 or ProGen2). Then, train on one split and test on the split with seqs with low seqID. Ideally, these seqs should not be homologous, IMO.
2/9 First, these questions were inspired by some comments of Leonardo Castorina about this this quote of Jumper
youtu.be/P_fHJIYENdI?...
But also (and mainly), in this answer by Ilya Sutskever at the last NeurIPS about the generalization capabilities of reasoning models
youtu.be/1yvBqasHLZs?...
youtu.be/P_fHJIYENdI?...
But also (and mainly), in this answer by Ilya Sutskever at the last NeurIPS about the generalization capabilities of reasoning models
youtu.be/1yvBqasHLZs?...


July 9, 2025 at 5:36 PM
2/9 First, these questions were inspired by some comments of Leonardo Castorina about this this quote of Jumper
youtu.be/P_fHJIYENdI?...
But also (and mainly), in this answer by Ilya Sutskever at the last NeurIPS about the generalization capabilities of reasoning models
youtu.be/1yvBqasHLZs?...
youtu.be/P_fHJIYENdI?...
But also (and mainly), in this answer by Ilya Sutskever at the last NeurIPS about the generalization capabilities of reasoning models
youtu.be/1yvBqasHLZs?...
🧵 1/9 I want to share some (inconclusive) thoughts about the generalization capabilities of protein language models under this main question: ¿what does it mean IN distribution generalization and what does it mean OUT of distribution generalization in (protein) biology?
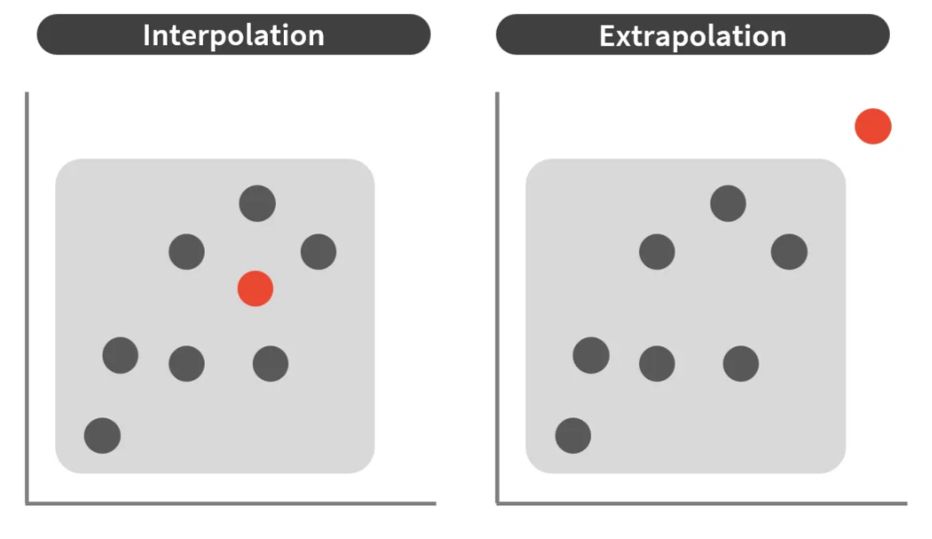
July 9, 2025 at 5:36 PM
🧵 1/9 I want to share some (inconclusive) thoughts about the generalization capabilities of protein language models under this main question: ¿what does it mean IN distribution generalization and what does it mean OUT of distribution generalization in (protein) biology?