



Melé Lab
@melelab.bsky.social
We are the Melé Lab, the Transcriptomics and Functional Genomics Lab (TFGL) at the Barcelona Supercomputing Center (BSC).
Account by lab members
Account by lab members
Our study reveals that menopause is a biological inflection point that initiates divergent aging trajectories across reproductive tissues.
Aging is asynchronous — and that’s a roadmap to smarter, more targeted interventions.
🔗 www.biorxiv.org/content/10.1...
8/8🧵
Aging is asynchronous — and that’s a roadmap to smarter, more targeted interventions.
🔗 www.biorxiv.org/content/10.1...
8/8🧵
www.biorxiv.org
May 23, 2025 at 12:19 PM
Our study reveals that menopause is a biological inflection point that initiates divergent aging trajectories across reproductive tissues.
Aging is asynchronous — and that’s a roadmap to smarter, more targeted interventions.
🔗 www.biorxiv.org/content/10.1...
8/8🧵
Aging is asynchronous — and that’s a roadmap to smarter, more targeted interventions.
🔗 www.biorxiv.org/content/10.1...
8/8🧵
Age-associated genes across uterus, myometrium & ovary overlap with genes from large GWAS or WES studies for:
- Menopause timing
- Age at Menarche
- Pelvic organ prolapse
The uterus isn’t just a passive player—it might hold the keys to predicting and preventing post-menopausal disorders.
7/8🧵
- Menopause timing
- Age at Menarche
- Pelvic organ prolapse
The uterus isn’t just a passive player—it might hold the keys to predicting and preventing post-menopausal disorders.
7/8🧵

May 23, 2025 at 12:19 PM
Age-associated genes across uterus, myometrium & ovary overlap with genes from large GWAS or WES studies for:
- Menopause timing
- Age at Menarche
- Pelvic organ prolapse
The uterus isn’t just a passive player—it might hold the keys to predicting and preventing post-menopausal disorders.
7/8🧵
- Menopause timing
- Age at Menarche
- Pelvic organ prolapse
The uterus isn’t just a passive player—it might hold the keys to predicting and preventing post-menopausal disorders.
7/8🧵
💡 Bulk RNA-seq isn’t tissue-specific.
But paired with histology, it can be.
We uncovered tissue-specific aging pathways:
🔻 Myometrium: ↓ ECM & muscle genes
🔺 Vaginal epithelium: ↓ epithelial +↑ immune genes
🔻 Ovarian cortex: ↓ angiogenesis
Organs don’t age as one.
6/8 🧵
But paired with histology, it can be.
We uncovered tissue-specific aging pathways:
🔻 Myometrium: ↓ ECM & muscle genes
🔺 Vaginal epithelium: ↓ epithelial +↑ immune genes
🔻 Ovarian cortex: ↓ angiogenesis
Organs don’t age as one.
6/8 🧵

May 23, 2025 at 12:19 PM
💡 Bulk RNA-seq isn’t tissue-specific.
But paired with histology, it can be.
We uncovered tissue-specific aging pathways:
🔻 Myometrium: ↓ ECM & muscle genes
🔺 Vaginal epithelium: ↓ epithelial +↑ immune genes
🔻 Ovarian cortex: ↓ angiogenesis
Organs don’t age as one.
6/8 🧵
But paired with histology, it can be.
We uncovered tissue-specific aging pathways:
🔻 Myometrium: ↓ ECM & muscle genes
🔺 Vaginal epithelium: ↓ epithelial +↑ immune genes
🔻 Ovarian cortex: ↓ angiogenesis
Organs don’t age as one.
6/8 🧵
The vaginal epithelium showed a menopause-timed crash in thickness.
This is more than known thinning: we saw extensive histological remodeling and sharp trajectory shifts at ~51 yrs.
❗Histology beats gene expression at capturing this transition.
5/8🧵
This is more than known thinning: we saw extensive histological remodeling and sharp trajectory shifts at ~51 yrs.
❗Histology beats gene expression at capturing this transition.
5/8🧵

May 23, 2025 at 12:19 PM
The vaginal epithelium showed a menopause-timed crash in thickness.
This is more than known thinning: we saw extensive histological remodeling and sharp trajectory shifts at ~51 yrs.
❗Histology beats gene expression at capturing this transition.
5/8🧵
This is more than known thinning: we saw extensive histological remodeling and sharp trajectory shifts at ~51 yrs.
❗Histology beats gene expression at capturing this transition.
5/8🧵
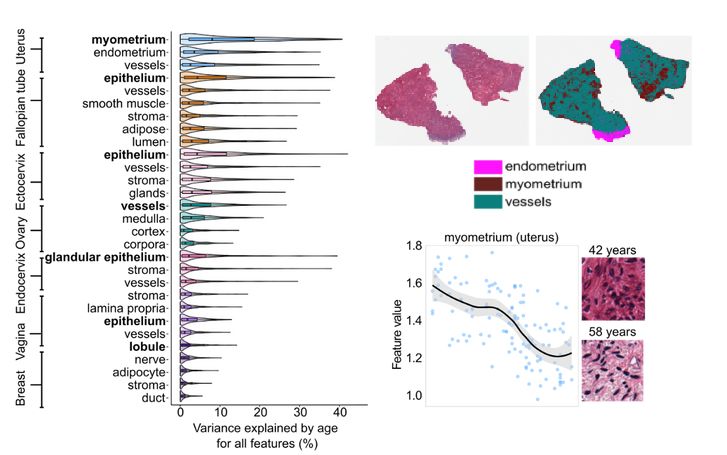
We zoomed in further.
Segmenting tissues with a vision transformer revealed that the myometrium (uterine muscle) is the most age-sensitive tissue.
Its histology transforms —collagen builds up, structure changes— with aging.
4/8🧵
Segmenting tissues with a vision transformer revealed that the myometrium (uterine muscle) is the most age-sensitive tissue.
Its histology transforms —collagen builds up, structure changes— with aging.
4/8🧵
May 23, 2025 at 12:19 PM
We zoomed in further.
Segmenting tissues with a vision transformer revealed that the myometrium (uterine muscle) is the most age-sensitive tissue.
Its histology transforms —collagen builds up, structure changes— with aging.
4/8🧵
Segmenting tissues with a vision transformer revealed that the myometrium (uterine muscle) is the most age-sensitive tissue.
Its histology transforms —collagen builds up, structure changes— with aging.
4/8🧵
Using CNNs, we trained aging classifiers per organ. Result?
📈 Ovaries & vagina show gradual aging
⚡️ Uterus shows abrupt transition around age 51—right at menopause!
Transcriptomic data mirrored this: gene expression shifts sharply in the uterus but not in the ovary or vagina.
3/8🧵
📈 Ovaries & vagina show gradual aging
⚡️ Uterus shows abrupt transition around age 51—right at menopause!
Transcriptomic data mirrored this: gene expression shifts sharply in the uterus but not in the ovary or vagina.
3/8🧵

May 23, 2025 at 12:19 PM
Using CNNs, we trained aging classifiers per organ. Result?
📈 Ovaries & vagina show gradual aging
⚡️ Uterus shows abrupt transition around age 51—right at menopause!
Transcriptomic data mirrored this: gene expression shifts sharply in the uterus but not in the ovary or vagina.
3/8🧵
📈 Ovaries & vagina show gradual aging
⚡️ Uterus shows abrupt transition around age 51—right at menopause!
Transcriptomic data mirrored this: gene expression shifts sharply in the uterus but not in the ovary or vagina.
3/8🧵
We combined:
🖼️ 1,112 histological images
🧬 659 RNA-seq samples
🧠 Deep learning + vision transformers
📊 Multi-omics integration
All from 304 women aged 20-70 (GTEx dataset).
Across 7 organs: uterus, ovary, vagina, breast, ectocervix, fallopian tubes, and endocervix.
2/8🧵
🖼️ 1,112 histological images
🧬 659 RNA-seq samples
🧠 Deep learning + vision transformers
📊 Multi-omics integration
All from 304 women aged 20-70 (GTEx dataset).
Across 7 organs: uterus, ovary, vagina, breast, ectocervix, fallopian tubes, and endocervix.
2/8🧵

May 23, 2025 at 12:19 PM
We combined:
🖼️ 1,112 histological images
🧬 659 RNA-seq samples
🧠 Deep learning + vision transformers
📊 Multi-omics integration
All from 304 women aged 20-70 (GTEx dataset).
Across 7 organs: uterus, ovary, vagina, breast, ectocervix, fallopian tubes, and endocervix.
2/8🧵
🖼️ 1,112 histological images
🧬 659 RNA-seq samples
🧠 Deep learning + vision transformers
📊 Multi-omics integration
All from 304 women aged 20-70 (GTEx dataset).
Across 7 organs: uterus, ovary, vagina, breast, ectocervix, fallopian tubes, and endocervix.
2/8🧵
We want to thank everyone 👩🔬 involved in this work: @pclavell.bsky.social, @fairlie.bsky.social, @scarbonells.bsky.social, @fdegalez.bsky.social, Carme Arnan, @oliveroswinona.bsky.social, @rodericguigo.bsky.social, @martamele.bsky.social as well as the institutions: @bsc-cns.bsky.social, @crg.eu ✨
March 20, 2025 at 1:20 PM
We want to thank everyone 👩🔬 involved in this work: @pclavell.bsky.social, @fairlie.bsky.social, @scarbonells.bsky.social, @fdegalez.bsky.social, Carme Arnan, @oliveroswinona.bsky.social, @rodericguigo.bsky.social, @martamele.bsky.social as well as the institutions: @bsc-cns.bsky.social, @crg.eu ✨
🟢Overall, our work suggests that to overcome gene annotation ancestry biases we require:
🥇long-read sequencing of transcriptomes from diverse human populations
🥈the development of transcript discovery tools for the pangenome @humanpangenome.bsky.social to bypass reference biases
Preprint ⬇️⬇️⬇️
13
🥇long-read sequencing of transcriptomes from diverse human populations
🥈the development of transcript discovery tools for the pangenome @humanpangenome.bsky.social to bypass reference biases
Preprint ⬇️⬇️⬇️
13

Long-read transcriptomics of a diverse human cohort reveals widespread ancestry bias in gene annotation
Accurate gene annotations are fundamental for interpreting genetic variation, cellular function, and disease mechanisms. However, current human gene annotations are largely derived from transcriptomic...
www.biorxiv.org
March 20, 2025 at 1:20 PM
🟢Overall, our work suggests that to overcome gene annotation ancestry biases we require:
🥇long-read sequencing of transcriptomes from diverse human populations
🥈the development of transcript discovery tools for the pangenome @humanpangenome.bsky.social to bypass reference biases
Preprint ⬇️⬇️⬇️
13
🥇long-read sequencing of transcriptomes from diverse human populations
🥈the development of transcript discovery tools for the pangenome @humanpangenome.bsky.social to bypass reference biases
Preprint ⬇️⬇️⬇️
13
🔴We saw that 🧬👤 full personal genome assemblies:
1️⃣ enable the discovery of novel transcripts that can not be found with GRCh38
2️⃣ contain regions not found in GRCh38 that harbor potentially novel genes (albeit at very low densities compared to the rest of the genome)
12
1️⃣ enable the discovery of novel transcripts that can not be found with GRCh38
2️⃣ contain regions not found in GRCh38 that harbor potentially novel genes (albeit at very low densities compared to the rest of the genome)
12

March 20, 2025 at 1:20 PM
🔴We saw that 🧬👤 full personal genome assemblies:
1️⃣ enable the discovery of novel transcripts that can not be found with GRCh38
2️⃣ contain regions not found in GRCh38 that harbor potentially novel genes (albeit at very low densities compared to the rest of the genome)
12
1️⃣ enable the discovery of novel transcripts that can not be found with GRCh38
2️⃣ contain regions not found in GRCh38 that harbor potentially novel genes (albeit at very low densities compared to the rest of the genome)
12
🔴This shows that the genome assembly of choice can potentially hide 🙈 transcripts due to genetic diversity.
To further explore the impact of *ALL* genetic diversity on transcript discovery, we did similar analyses comparing GRCh38 with 🧬👤 full personal genome assemblies ⬇️
11
To further explore the impact of *ALL* genetic diversity on transcript discovery, we did similar analyses comparing GRCh38 with 🧬👤 full personal genome assemblies ⬇️
11
March 20, 2025 at 1:20 PM
🔴This shows that the genome assembly of choice can potentially hide 🙈 transcripts due to genetic diversity.
To further explore the impact of *ALL* genetic diversity on transcript discovery, we did similar analyses comparing GRCh38 with 🧬👤 full personal genome assemblies ⬇️
11
To further explore the impact of *ALL* genetic diversity on transcript discovery, we did similar analyses comparing GRCh38 with 🧬👤 full personal genome assemblies ⬇️
11
💥We find that personalized-GRCh38s enable the discovery of >3% more novel transcripts than using GRCh38.
🌍More importantly, this increase is uneven between populations, with African individuals benefiting most (LWK, YRI).
10
🌍More importantly, this increase is uneven between populations, with African individuals benefiting most (LWK, YRI).
10

March 20, 2025 at 1:20 PM
💥We find that personalized-GRCh38s enable the discovery of >3% more novel transcripts than using GRCh38.
🌍More importantly, this increase is uneven between populations, with African individuals benefiting most (LWK, YRI).
10
🌍More importantly, this increase is uneven between populations, with African individuals benefiting most (LWK, YRI).
10
💡Our hypothesis was that genetic variants differing from the reference genome sequence could interfere in the transcript discovery process.
So we compared transcript discovery when using GRCh38 and modified (👤personalized) versions of GRCh38 containing each sample's SNPs ⬇️
9
So we compared transcript discovery when using GRCh38 and modified (👤personalized) versions of GRCh38 containing each sample's SNPs ⬇️
9
March 20, 2025 at 1:20 PM
💡Our hypothesis was that genetic variants differing from the reference genome sequence could interfere in the transcript discovery process.
So we compared transcript discovery when using GRCh38 and modified (👤personalized) versions of GRCh38 containing each sample's SNPs ⬇️
9
So we compared transcript discovery when using GRCh38 and modified (👤personalized) versions of GRCh38 containing each sample's SNPs ⬇️
9
So far, we’ve shown that reference gene annotations are European biased ✅
❓However, would it be enough to sequence RNA from genetically diverse cohorts to build truly representative gene annotations?
So we searched 🔎 for potential biases caused by using reference genome assemblies (GRCh38) ⬇️
8
❓However, would it be enough to sequence RNA from genetically diverse cohorts to build truly representative gene annotations?
So we searched 🔎 for potential biases caused by using reference genome assemblies (GRCh38) ⬇️
8
March 20, 2025 at 1:20 PM
So far, we’ve shown that reference gene annotations are European biased ✅
❓However, would it be enough to sequence RNA from genetically diverse cohorts to build truly representative gene annotations?
So we searched 🔎 for potential biases caused by using reference genome assemblies (GRCh38) ⬇️
8
❓However, would it be enough to sequence RNA from genetically diverse cohorts to build truly representative gene annotations?
So we searched 🔎 for potential biases caused by using reference genome assemblies (GRCh38) ⬇️
8
This is precisely what happens when linking 🧬 genetic variation to 📈 transcript expression ⬇️
🔴We show that a population-diverse gene annotation increases the discovery of associations between SNPs and transcript expression, especially in non-European populations 🌍
7
🔴We show that a population-diverse gene annotation increases the discovery of associations between SNPs and transcript expression, especially in non-European populations 🌍
7

March 20, 2025 at 1:20 PM
This is precisely what happens when linking 🧬 genetic variation to 📈 transcript expression ⬇️
🔴We show that a population-diverse gene annotation increases the discovery of associations between SNPs and transcript expression, especially in non-European populations 🌍
7
🔴We show that a population-diverse gene annotation increases the discovery of associations between SNPs and transcript expression, especially in non-European populations 🌍
7
❓Why is this important?
Gene annotations enable the interpretation of the human genome sequence by providing context on how the molecular actors (RNA/proteins) play a role to produce different phenotypes.
➡️If gene annotations are biased, so too might be our interpretation of the genome
6
Gene annotations enable the interpretation of the human genome sequence by providing context on how the molecular actors (RNA/proteins) play a role to produce different phenotypes.
➡️If gene annotations are biased, so too might be our interpretation of the genome
6
March 20, 2025 at 1:20 PM
❓Why is this important?
Gene annotations enable the interpretation of the human genome sequence by providing context on how the molecular actors (RNA/proteins) play a role to produce different phenotypes.
➡️If gene annotations are biased, so too might be our interpretation of the genome
6
Gene annotations enable the interpretation of the human genome sequence by providing context on how the molecular actors (RNA/proteins) play a role to produce different phenotypes.
➡️If gene annotations are biased, so too might be our interpretation of the genome
6
➡️This is a direct consequence of the systematic underrepresentation of non-European samples 🌍 throughout the -omics fields.
🔴In other words, the data used to build current gene annotations is not representative of global human genetic diversity.
5
🔴In other words, the data used to build current gene annotations is not representative of global human genetic diversity.
5
March 20, 2025 at 1:20 PM
➡️This is a direct consequence of the systematic underrepresentation of non-European samples 🌍 throughout the -omics fields.
🔴In other words, the data used to build current gene annotations is not representative of global human genetic diversity.
5
🔴In other words, the data used to build current gene annotations is not representative of global human genetic diversity.
5
In all samples, we found many transcripts that have never been seen before (~40k total) 👀
❗Interestingly, we find more novel transcripts in non-European ancestry samples than in European ones.
🔴This means that gene annotations systematically miss more transcripts from non-European individuals.
4
❗Interestingly, we find more novel transcripts in non-European ancestry samples than in European ones.
🔴This means that gene annotations systematically miss more transcripts from non-European individuals.
4
March 20, 2025 at 1:20 PM
In all samples, we found many transcripts that have never been seen before (~40k total) 👀
❗Interestingly, we find more novel transcripts in non-European ancestry samples than in European ones.
🔴This means that gene annotations systematically miss more transcripts from non-European individuals.
4
❗Interestingly, we find more novel transcripts in non-European ancestry samples than in European ones.
🔴This means that gene annotations systematically miss more transcripts from non-European individuals.
4
Together with @guigolab.bsky.social 🙌 we deeply sequenced RNA of 43 cell lines (LCL) 🧫 from 8 human populations 👥with distinct genetic ancestries 🌍 using ONT (@nanoporetech.com) to capture full-length mRNA and lncRNA molecules.
3
3

March 20, 2025 at 1:20 PM
Together with @guigolab.bsky.social 🙌 we deeply sequenced RNA of 43 cell lines (LCL) 🧫 from 8 human populations 👥with distinct genetic ancestries 🌍 using ONT (@nanoporetech.com) to capture full-length mRNA and lncRNA molecules.
3
3
⚠️Most transcriptomic data comes from European-descent individuals; implying that data used to build gene annotations does too. Furthermore, there are no long-read transcriptomic datasets that sample 🌍 diverse human populations
❓We wondered if this could bias reference gene annotations
2
❓We wondered if this could bias reference gene annotations
2
March 20, 2025 at 1:20 PM
⚠️Most transcriptomic data comes from European-descent individuals; implying that data used to build gene annotations does too. Furthermore, there are no long-read transcriptomic datasets that sample 🌍 diverse human populations
❓We wondered if this could bias reference gene annotations
2
❓We wondered if this could bias reference gene annotations
2