



Laurent Jacob
@laurentjacob.bsky.social
The decisions for LEGEND are out: legend2025.sciencesconf.org/data/book_le...
I'm really looking forward to hearing these 21 exciting presentations (and additional 30 posters) next December.
If you want to attend too, registration is open until October 17th through legend2025.sciencesconf.org
I'm really looking forward to hearing these 21 exciting presentations (and additional 30 posters) next December.
If you want to attend too, registration is open until October 17th through legend2025.sciencesconf.org
![Exploring the space of self-reproducing RNA using generative models, Martin Weigt
Exploring the archaic introgression landscape of admixed populations through
joint ancestry inference, Jazeps Medina Tretmanis [et al.]
Predicting natural variation in the yeast phenotypic landscape with machine
learning, Sakshi Khaiwal [et al.]
Phylodynamic modeling with unsupervised Bayesian neural networks, Marino
Gabriele [et al.]
Likelihood-free inference of phylogenetic tree posterior distributions, Luc Blas-
sel [et al.]
Generative continuous time model reveals epistatic signatures in protein evolu-
tion, Barrat-Charlaix Pierre
Neural posterior estimation for high-dimensional genomic data from complex pop-
ulation genetic models, Jiseon Min [et al.]
A differentiable model for detecting diversifying selection directly from alignments
in large-scale bacterial datasets, Leonie Lorenz [et al.]
Detecting interspecific positive selection using transformers, Charlotte West [et al.]
Predicting Multiple Sequence Alignment Uncertainty via Machine Learning, Lucia
Martin-Fernandez [et al.]
Graph Neural Networks for Likelihood-Free Inference in Diversification Mod-
els, Amélie Leroy [et al.]
Popformer: learning general signatures of genetic variation and natural selection
with a self-supervised transformer, Leon Zong [et al.]](https://cdn.bsky.app/img/feed_thumbnail/plain/did:plc:7x5c6ifrcznmg4mzhfuc2a6r/bafkreidur7bz43o2jhwr4gbt36awozawqthbkkfeszxjkxf4kvhyh7q6du@jpeg)
![PRIVET: PRIVacy metric based on Extreme value Theory, Antoine Szatkownik [et
al.]
Generative models for inferring the evolutionary history of the malaria vector
Anopheles gambiae, Amelia Eneli [et al.]
Language Models Outperform Supervised-Only Approaches for Conserved Ele-
ment Comprehension, Eyes Robson [et al.]
Identification and Classification of Orphan Genes, Spurious Orphan Genes, and
Conserved Genes from the human microbiome, Chen Chen
Neural Simulation-based inference of demography and selection, Francisco De
Borja Campuzano Jiménez [et al.]
Species Identification and aDNA Read Mapping Using k-mer Embeddings, Filip
Thor [et al.]
Contrastive Learning for Population Structure and Trait Prediction, Filip Thor [et
al.]
Protein and genomic language models chart a vast landscape of antiphage de-
fenses, Mordret Ernest
The Phylogenomics and Sparse Learning of Trait Innovations, Gaurav Diwan [et
al.]](https://cdn.bsky.app/img/feed_thumbnail/plain/did:plc:7x5c6ifrcznmg4mzhfuc2a6r/bafkreig4akadduemzbit67bv7jnr7x2ircwiriykabg2imuf3phy6hhube@jpeg)
October 8, 2025 at 11:04 AM
The decisions for LEGEND are out: legend2025.sciencesconf.org/data/book_le...
I'm really looking forward to hearing these 21 exciting presentations (and additional 30 posters) next December.
If you want to attend too, registration is open until October 17th through legend2025.sciencesconf.org
I'm really looking forward to hearing these 21 exciting presentations (and additional 30 posters) next December.
If you want to attend too, registration is open until October 17th through legend2025.sciencesconf.org
Come hear about the latest advances in the field and discuss your own work at Centre Paul Langevin in beautiful Aussois.

February 24, 2025 at 8:58 AM
Come hear about the latest advances in the field and discuss your own work at Centre Paul Langevin in beautiful Aussois.
Burak Yelmen from the University of Tartu will give a keynote presentation on "A perspective on generative neural networks in genomics with applications in synthetic data generation".

February 24, 2025 at 8:58 AM
Burak Yelmen from the University of Tartu will give a keynote presentation on "A perspective on generative neural networks in genomics with applications in synthetic data generation".
Claudia Solís-Lemus from the University of Wisconsin-Madison will give a keynote presentation on "The good, the bad and the ugly of deep learning in phylogenetic inference".

February 24, 2025 at 8:58 AM
Claudia Solís-Lemus from the University of Wisconsin-Madison will give a keynote presentation on "The good, the bad and the ugly of deep learning in phylogenetic inference".
Anne-Florence Bitbol from EPFL will give a keynote presentation on "Coevolution-aware language models".

February 24, 2025 at 8:58 AM
Anne-Florence Bitbol from EPFL will give a keynote presentation on "Coevolution-aware language models".
The next LEGEND conference on machine learning for evolutionary genomics will be in Aussois (French Alps) between December 8th and 12th.
Mark your calendars and make sure your best work is ready next September when the call for abstracts opens 🙂
legend2025.sciencesconf.org
Mark your calendars and make sure your best work is ready next September when the call for abstracts opens 🙂
legend2025.sciencesconf.org

February 24, 2025 at 8:58 AM
The next LEGEND conference on machine learning for evolutionary genomics will be in Aussois (French Alps) between December 8th and 12th.
Mark your calendars and make sure your best work is ready next September when the call for abstracts opens 🙂
legend2025.sciencesconf.org
Mark your calendars and make sure your best work is ready next September when the call for abstracts opens 🙂
legend2025.sciencesconf.org
All this work was done by Luca Nesterenko and
@lblassel.bsky.social , assisted by P. Veber, Bastien Boussau
and myself.
The code and data are available at github.com/lucanest/Phy...
Please share if you find this interesting, and we welcome your feedback :)
@lblassel.bsky.social , assisted by P. Veber, Bastien Boussau
and myself.
The code and data are available at github.com/lucanest/Phy...
Please share if you find this interesting, and we welcome your feedback :)
June 24, 2024 at 8:35 AM
All this work was done by Luca Nesterenko and
@lblassel.bsky.social , assisted by P. Veber, Bastien Boussau
and myself.
The code and data are available at github.com/lucanest/Phy...
Please share if you find this interesting, and we welcome your feedback :)
@lblassel.bsky.social , assisted by P. Veber, Bastien Boussau
and myself.
The code and data are available at github.com/lucanest/Phy...
Please share if you find this interesting, and we welcome your feedback :)
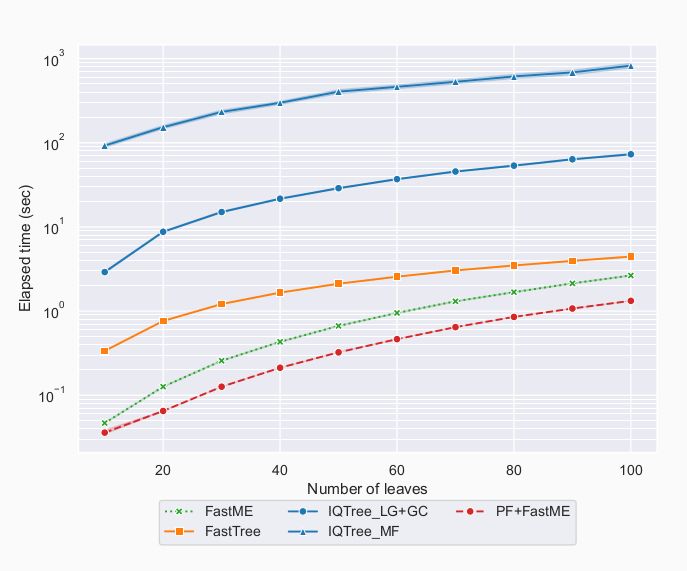
In all these experiments, and regardless of model complexity, Phyloformer run on a GPU was the fastest method.
About two orders of magnitude faster than IQtree, and even twice faster than FastME.
About two orders of magnitude faster than IQtree, and even twice faster than FastME.
June 24, 2024 at 8:33 AM
In all these experiments, and regardless of model complexity, Phyloformer run on a GPU was the fastest method.
About two orders of magnitude faster than IQtree, and even twice faster than FastME.
About two orders of magnitude faster than IQtree, and even twice faster than FastME.
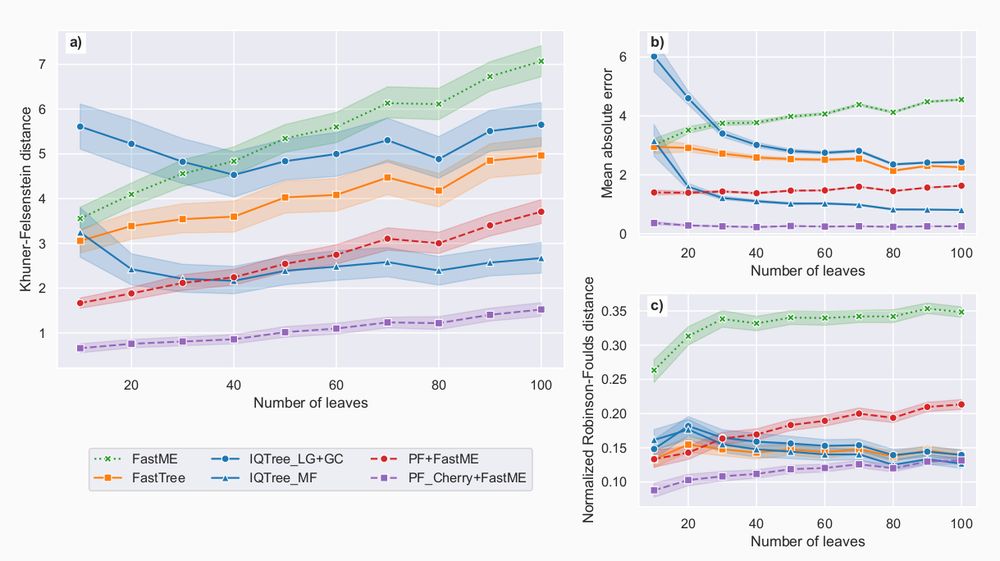
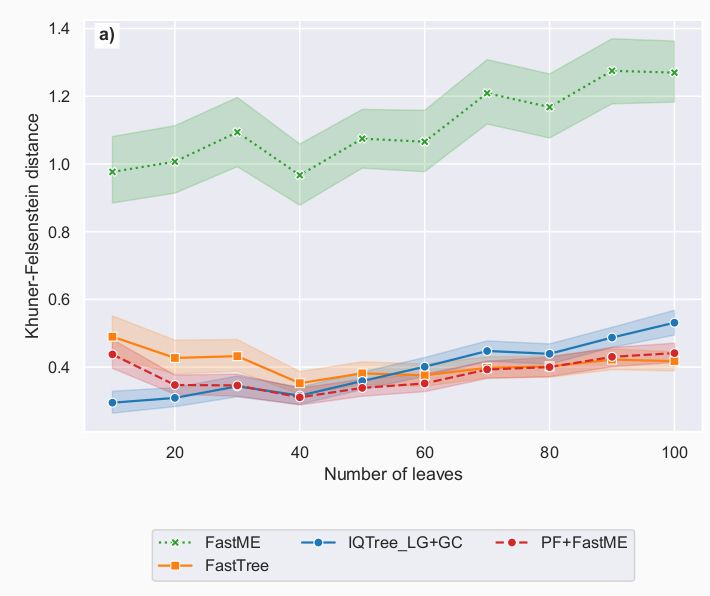
We then trained Phyloformer under a more realistic model, accounting for co-evolution.
It outperformed all other methods, including IQTree/FastTree, on all metrics.
It outperformed all other methods, including IQTree/FastTree, on all metrics.
June 24, 2024 at 8:32 AM
We then trained Phyloformer under a more realistic model, accounting for co-evolution.
It outperformed all other methods, including IQTree/FastTree, on all metrics.
It outperformed all other methods, including IQTree/FastTree, on all metrics.
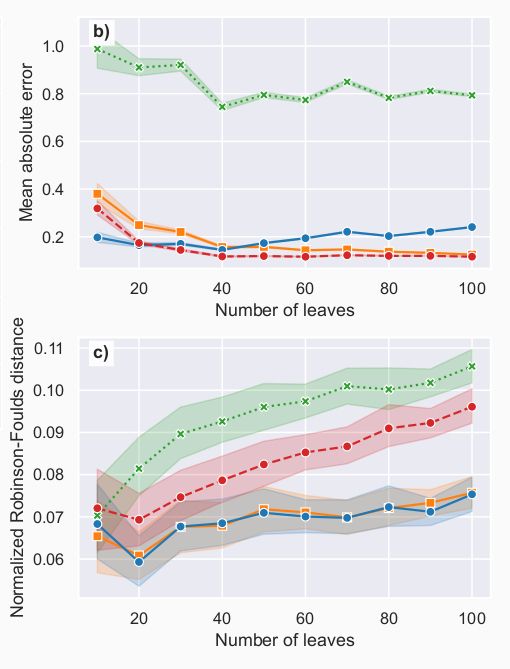
More precisely, Phyloformer was very good at predicting distances, and on the Kuhner-Felsenstein metric accounting for both topology and branch lengths.
Looking at the topology only (Robinson-Foulds metric), it performed less well than IQTree/FastTree, but better than FastME.
Looking at the topology only (Robinson-Foulds metric), it performed less well than IQTree/FastTree, but better than FastME.
June 24, 2024 at 8:32 AM
More precisely, Phyloformer was very good at predicting distances, and on the Kuhner-Felsenstein metric accounting for both topology and branch lengths.
Looking at the topology only (Robinson-Foulds metric), it performed less well than IQTree/FastTree, but better than FastME.
Looking at the topology only (Robinson-Foulds metric), it performed less well than IQTree/FastTree, but better than FastME.
We first trained Phyloformer to perform inference under LG, a common model under which likelihood computation is possible.
It performed much better than FastME (distance method), on par with maximum likelihood approaches (IQTree, FastTree).
It performed much better than FastME (distance method), on par with maximum likelihood approaches (IQTree, FastTree).
June 24, 2024 at 8:31 AM
We first trained Phyloformer to perform inference under LG, a common model under which likelihood computation is possible.
It performed much better than FastME (distance method), on par with maximum likelihood approaches (IQTree, FastTree).
It performed much better than FastME (distance method), on par with maximum likelihood approaches (IQTree, FastTree).
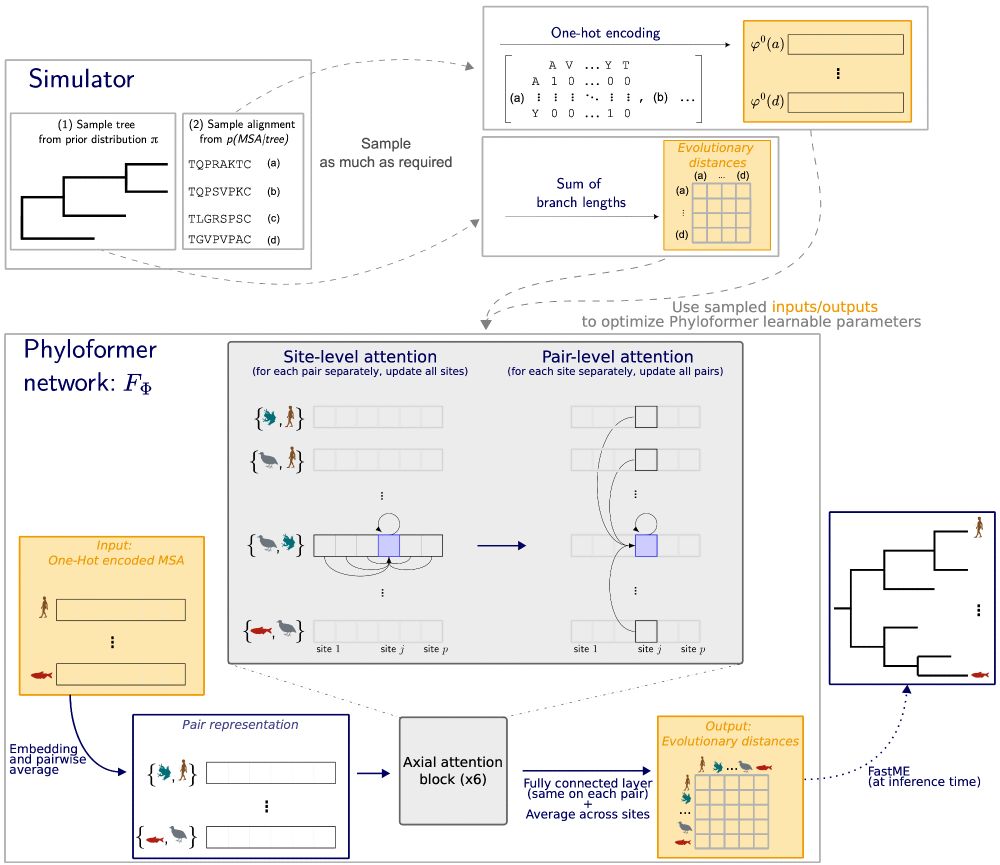
Phyloformer uses self-attention to progressively share information among and between sequences.
This choice makes our function invariant to the order of the input sequences (any order yields the same output phylogeny).
This choice makes our function invariant to the order of the input sequences (any order yields the same output phylogeny).
June 24, 2024 at 8:30 AM
Phyloformer uses self-attention to progressively share information among and between sequences.
This choice makes our function invariant to the order of the input sequences (any order yields the same output phylogeny).
This choice makes our function invariant to the order of the input sequences (any order yields the same output phylogeny).
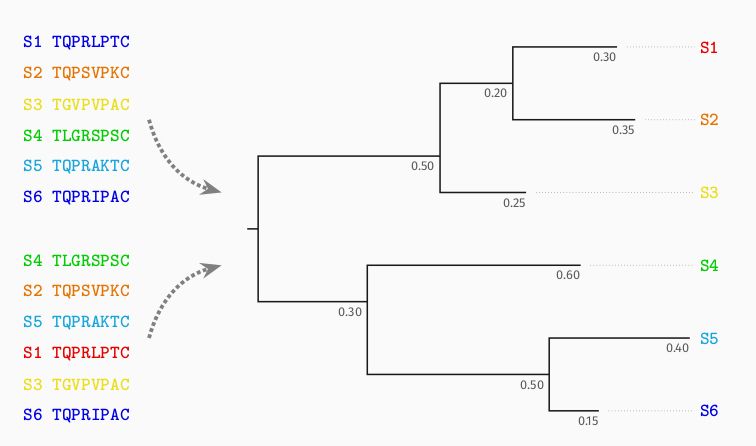
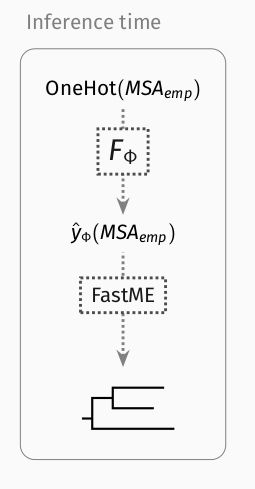
Once trained, Phyloformer provides estimates of all evolutionary distances given the sequences.
But each of these distance estimates is informed by the entire set of sequence, not just the corresponding pair!
We then pass them to FastME, a distance method, to obtain a tree.
But each of these distance estimates is informed by the entire set of sequence, not just the corresponding pair!
We then pass them to FastME, a distance method, to obtain a tree.
June 24, 2024 at 8:29 AM
Once trained, Phyloformer provides estimates of all evolutionary distances given the sequences.
But each of these distance estimates is informed by the entire set of sequence, not just the corresponding pair!
We then pass them to FastME, a distance method, to obtain a tree.
But each of these distance estimates is informed by the entire set of sequence, not just the corresponding pair!
We then pass them to FastME, a distance method, to obtain a tree.
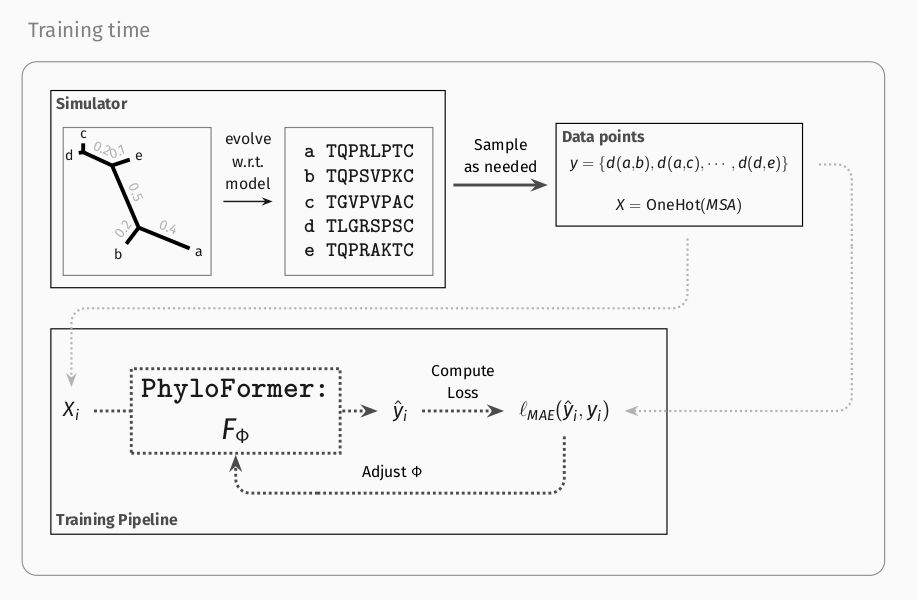
Phyloformer is a learnable function. Its input is a set of sequences, its output is their phylogeny, represented by evolutionary distances between all pairs of sequences.
We optimize this function on a large number of (phylogeny, sequences) sampled from the probabilistic model.
We optimize this function on a large number of (phylogeny, sequences) sampled from the probabilistic model.
June 24, 2024 at 8:28 AM
Phyloformer is a learnable function. Its input is a set of sequences, its output is their phylogeny, represented by evolutionary distances between all pairs of sequences.
We optimize this function on a large number of (phylogeny, sequences) sampled from the probabilistic model.
We optimize this function on a large number of (phylogeny, sequences) sampled from the probabilistic model.
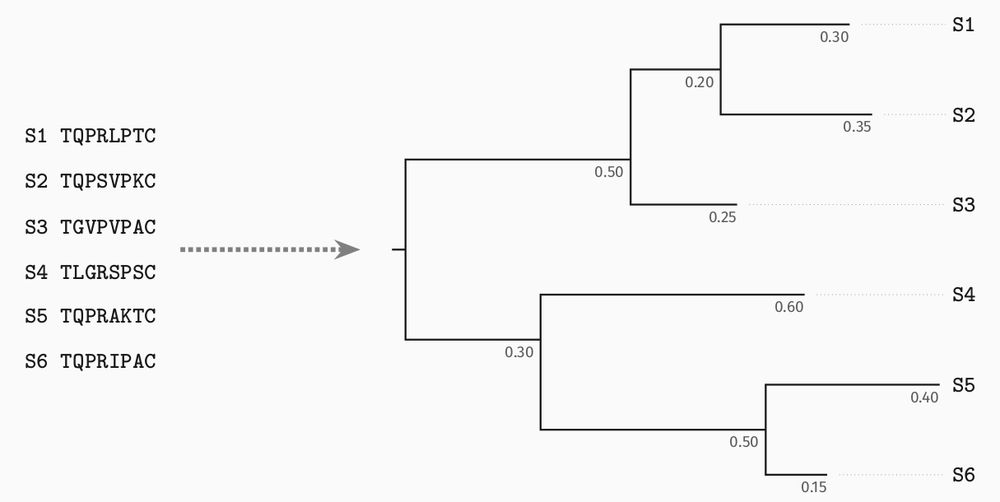
This is where likelihood-free/simulation-based inference comes into play.
Sampling trees and sequences under a probabilistic model is possible under much more complex models, for which likelihood computations would be prohibitive.
It's an alternative way to access the model.
Sampling trees and sequences under a probabilistic model is possible under much more complex models, for which likelihood computations would be prohibitive.
It's an alternative way to access the model.
June 24, 2024 at 8:27 AM
This is where likelihood-free/simulation-based inference comes into play.
Sampling trees and sequences under a probabilistic model is possible under much more complex models, for which likelihood computations would be prohibitive.
It's an alternative way to access the model.
Sampling trees and sequences under a probabilistic model is possible under much more complex models, for which likelihood computations would be prohibitive.
It's an alternative way to access the model.
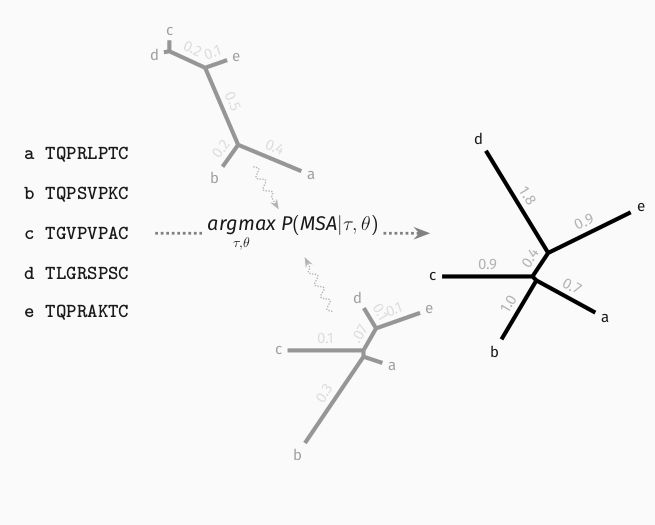
Maximum likelihood approaches on the other hand search for the most likely tree jointly over all sequences.
This makes them accurate but slow. It also restricts these approaches to simplistic models under which likelihood computations are fast enough.
This makes them accurate but slow. It also restricts these approaches to simplistic models under which likelihood computations are fast enough.
June 24, 2024 at 8:26 AM
Maximum likelihood approaches on the other hand search for the most likely tree jointly over all sequences.
This makes them accurate but slow. It also restricts these approaches to simplistic models under which likelihood computations are fast enough.
This makes them accurate but slow. It also restricts these approaches to simplistic models under which likelihood computations are fast enough.
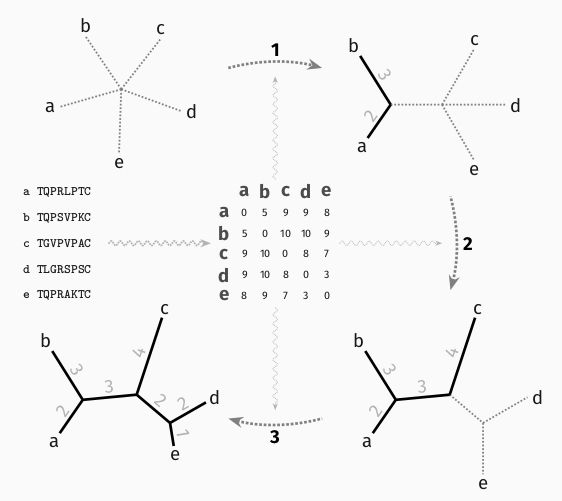
Knowing the evolutionary distances (sum of branch lengths) between all pairs of sequences is enough to recover the tree, by hierarchical clustering.
Distance methods rely on this idea, with estimates from pairs of sequences taken separately. This makes them fast but inaccurate.
Distance methods rely on this idea, with estimates from pairs of sequences taken separately. This makes them fast but inaccurate.
June 24, 2024 at 8:25 AM
Knowing the evolutionary distances (sum of branch lengths) between all pairs of sequences is enough to recover the tree, by hierarchical clustering.
Distance methods rely on this idea, with estimates from pairs of sequences taken separately. This makes them fast but inaccurate.
Distance methods rely on this idea, with estimates from pairs of sequences taken separately. This makes them fast but inaccurate.
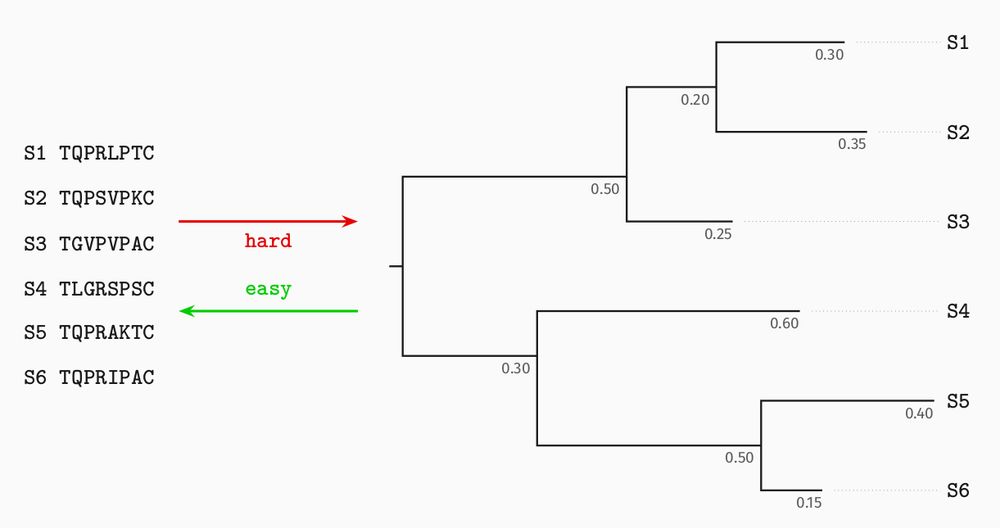
Phylogenetic trees describe how related sequences (at the leaves) evolved from a common ancestor. Internal nodes are successive ancestral sequences.
In probabilistic models, branch lengths represent an expected number of substitutions between the sequences at the two ends.
In probabilistic models, branch lengths represent an expected number of substitutions between the sequences at the two ends.
June 24, 2024 at 8:25 AM
Phylogenetic trees describe how related sequences (at the leaves) evolved from a common ancestor. Internal nodes are successive ancestral sequences.
In probabilistic models, branch lengths represent an expected number of substitutions between the sequences at the two ends.
In probabilistic models, branch lengths represent an expected number of substitutions between the sequences at the two ends.
We just released a preprint for Phyloformer, a likelihood-free inference method for phylogenetic reconstruction: biorxiv.org/content/10.1...
Faster than distance methods like neighbor joining, it outperforms maximum likelihood methods under complex models of sequence evolution.
🧵
Faster than distance methods like neighbor joining, it outperforms maximum likelihood methods under complex models of sequence evolution.
🧵

June 24, 2024 at 8:24 AM
We just released a preprint for Phyloformer, a likelihood-free inference method for phylogenetic reconstruction: biorxiv.org/content/10.1...
Faster than distance methods like neighbor joining, it outperforms maximum likelihood methods under complex models of sequence evolution.
🧵
Faster than distance methods like neighbor joining, it outperforms maximum likelihood methods under complex models of sequence evolution.
🧵